黄斑ジストロフィー(指定難病301)
○ 概要
1.概要
黄斑ジストロフィー(macular dystrophy)は眼底の黄斑部に両眼性、進行性の病変を呈する遺伝性疾患の総称である。一般にジストロフィー(dystrophy)とは、非炎症性、進行性の栄養上あるいは代謝上の異常を意味する。すなわち、黄斑ジストロフィーとは、何らかの遺伝子異常によって黄斑部の機能障害を来す一群の疾患であると考えられている。黄斑ジストロフィーの診断には、(1)基本的に両眼性である。(2)家族性、遺伝性の疾患である。(3)なんら外因が加わることなしに発生する。(4)他覚的検査所見、自覚的機能検査所見、いずれからみても徐々に進行する。これらの4つの項目を満たす(厚生省特性疾患網膜脈絡膜萎縮症調査研究班 黄斑ジストロフィー診断の手引きを参照)。なお黄斑ジストロフィーとは狭義には病変が黄斑部に限局した状態を指すが、多くの病型において病変は黄斑部から後極部に広がり、ときに周辺部網膜まで及ぶことがある。
2.原因
黄斑ジストロフィーのいくつかの病型については原因遺伝子が報告されているが、原因遺伝子が不明のものもある(表1)。各黄斑ジストロフィーの詳細な発症原因は不明のものも多い。
3.症状
徐々に進行する両眼の視力低下、色覚異常、中心視野異常、羞明。自覚症状の出現時期は、幼児期から中高年期までと幅広い。
4.治療法
治療法はない。
5.予後
次第に視力低下が進行し、矯正視力が0.1以下となることも多い。このため、特に書字・識字において著しい困難を生じるが、周辺視野は保たれるため完全な失明には至らない*。
*錐体-杆体ジストロフィーでは進行期には著明な周辺視野狭窄が見られることがある。
○ 要件の判定に必要な事項
1. 患者数
1,000名
2. 発病の機構
不明
3. 効果的な治療方法
未確立
4. 長期の療養
必要(進行性に視力低下を認める。)
5. 診断基準
あり(日本眼科学会で承認予定の診断基準あり)
6. 重症度分類
良好な方の眼の矯正視力が0.3未満の者を対象とする。
○ 情報提供元
日本眼科学会
<診断基準>
Definiteを対象とする。
A.症状
両眼視力低下(急性の視力低下は除外する。) 尚、視力低下の程度は問わない。
B.検査所見
① 眼底写真:両眼黄斑部の対称性の萎縮性病変、黄斑分離、あるいは沈着物
② 蛍光眼底造影(FA)又は眼底自発蛍光:病巣に一致した異常蛍光
③ 電気生理学的検討:
・全視野ERG(とくに錐体系)の反応減弱
・多局所及び黄斑局所ERGの反応減弱
・EOGのL/D比の低下
④ 光干渉断層計(OCT):病巣部における網膜の形態異常
C.鑑別診断
以下の疾患を鑑別する。
・ 薬物による視力低下
クロロキン、ハイドロオキシクロロキン、ティオリダジン、タモキシフェン等
・ 外傷性(あるいは近視性)網脈絡膜萎縮
・ 後天性網脈絡膜疾患 (中心性漿液性脈絡網膜症(CSC)、急性帯状潜在性網膜外層症(AZOOR)、MEWDS等)
・ 先天性コロボーマ、先天性黄斑低形成
・ 加齢黄斑変性萎縮型:年齢50歳以上の症例において黄斑に地図状萎縮が見られる。地図状萎縮は、1)直径250µm以上、2)円形、卵円形、房状又は地図状の形態、3)境界鮮明、4)網膜色素上皮の低色素又は脱色素変化、5)脈絡膜中大血管が明瞭に透見可能の全てを満たし、軟性ドルーゼン、reticular pseudodrusen、色素沈着を伴うことがある。
・ 続発性黄斑変性:黄斑疾患の既往がある。萎縮病変は両眼非対称性
D.家族歴
<診断のカテゴリー>
Definite:
・ A項目を満たし、かつB項目のうち3項目以上を満たし、Cの鑑別すべき疾患を除外したもの
・ B項目の4項目を全て満たし、Cの鑑別すべき疾患を除外する、かつ現在視力が良好でも、黄斑部萎縮の進行により将来視力が低下する可能性が高い。
・ 検査所見の特徴からそれぞれの病型の診断の要件を満たせばDefiniteとする。
・ Probableの項目を満たし、明らかな家族歴を満たせばDefiniteとする。
Probable:
B項目のうち2項目以上を満たし、Cの鑑別すべき疾患を除外したもの。
A項目あるいはB項目の1項目以上があり、C項目の鑑別すべきものを除外したもの。
<特異的な所見から診断が可能なもの>
(検査所見の特徴からそれぞれ以下の要件を満たした場合にDefiniteとする)
1.卵黄状黄斑ジストロフィー(ベスト病)
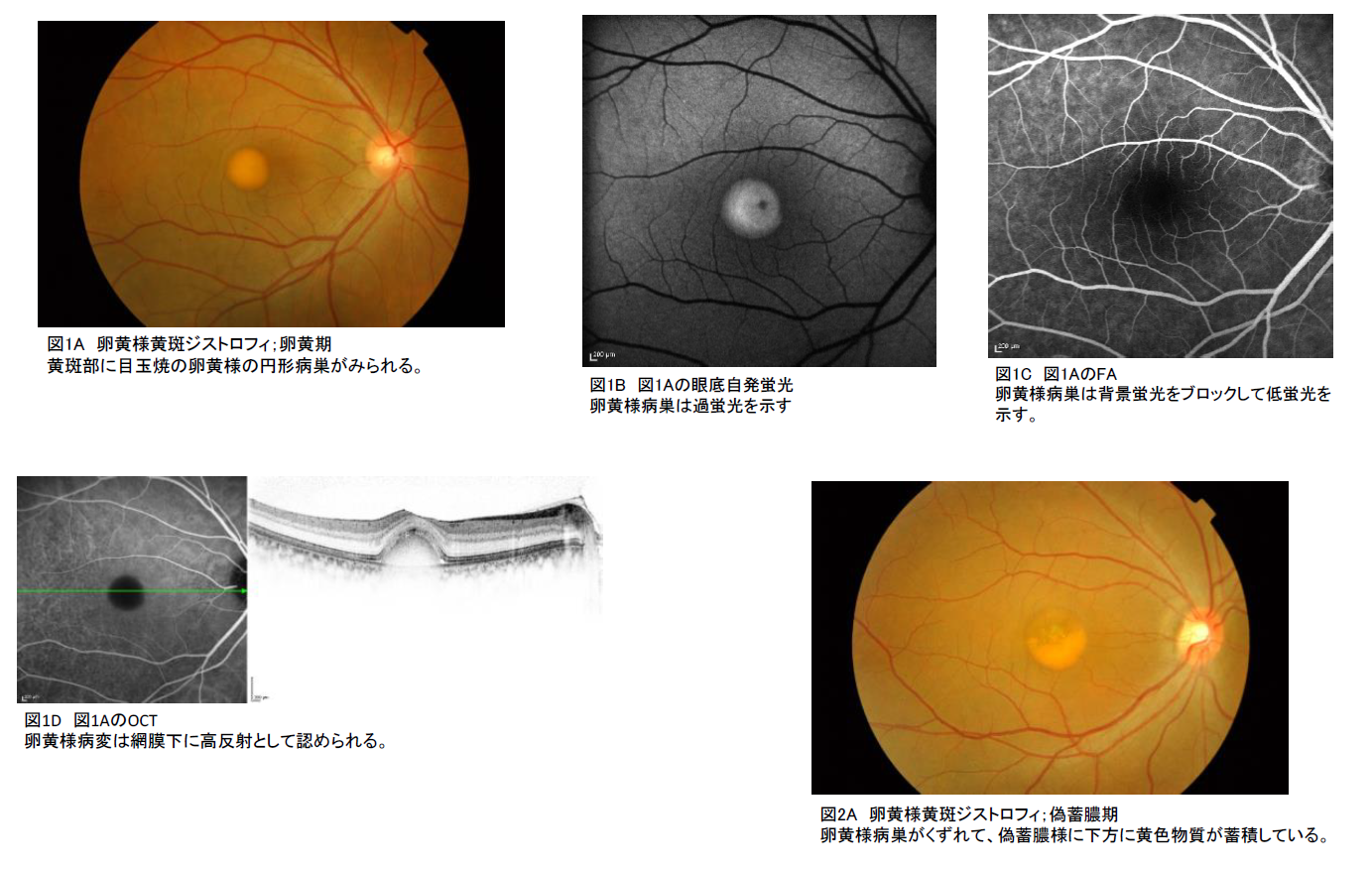
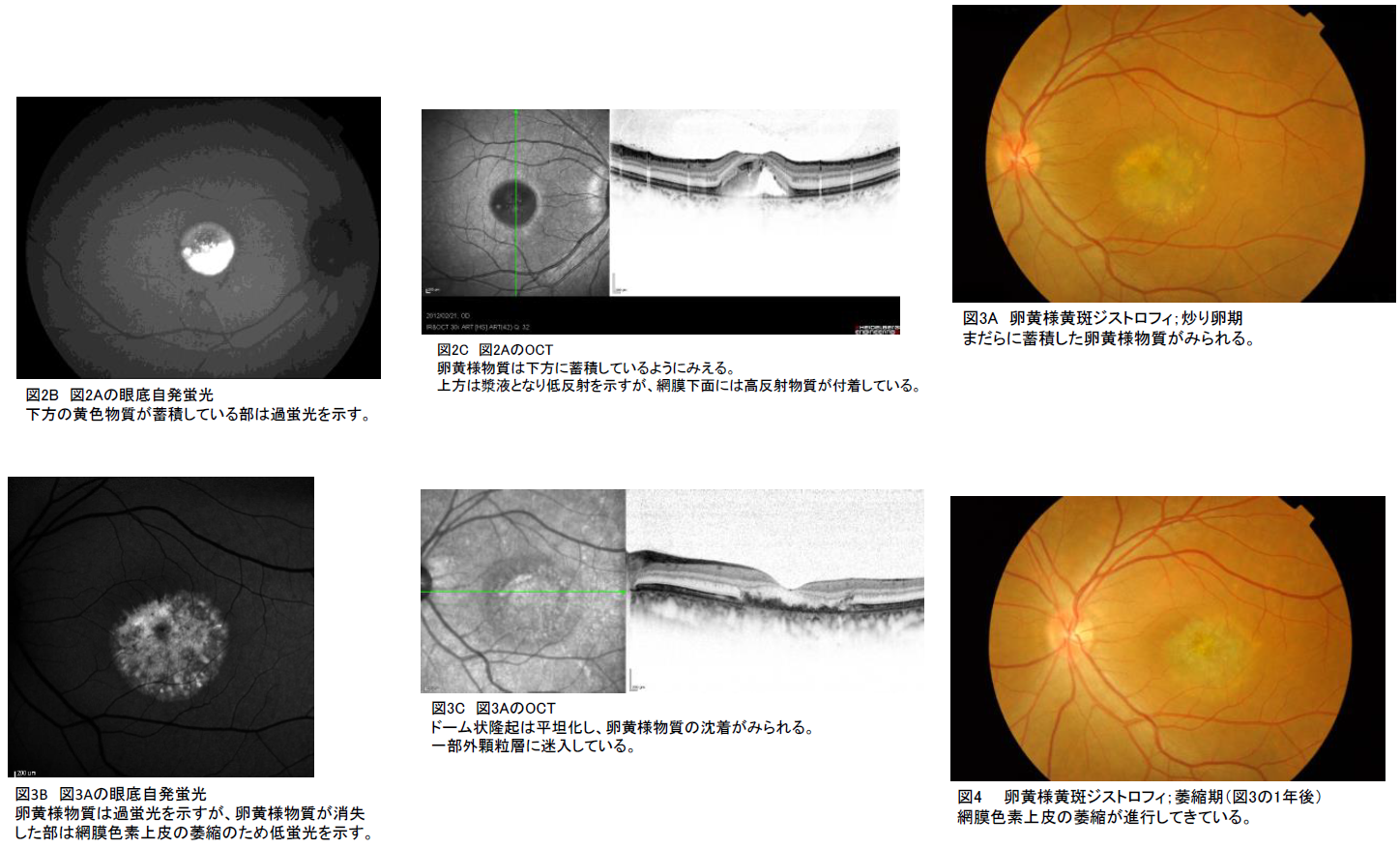
B-① 眼底写真(必須):卵黄様病巣(図1A)、偽蓄膿様病巣(図2A)、いり卵様病巣(図3A)を認める。
それぞれ図を参照。卵黄様物質が吸収すると非典型的な萎縮病巣になる(図4)。
B-② 眼底自発蛍光:卵黄様物質は過蛍光(図1B、図2B、図3B)を示す。
蛍光眼底造影(FA):卵黄様物質はブロックによる低蛍光(図1C)を示す。
B-③ 電気生理学的検討(必須):EOGはL/D比が低下する。
B-④ OCT:卵黄様黄斑物質は網膜下に貯留している(図1D、図2C、図3C)。
●診断の要件
診断はB-①眼底写真とB-②眼底自発蛍光あるいはFAとB-③電気生理学的検査で双方ともに上記の特徴を満たす場合に診断する。
2.Stargardt病
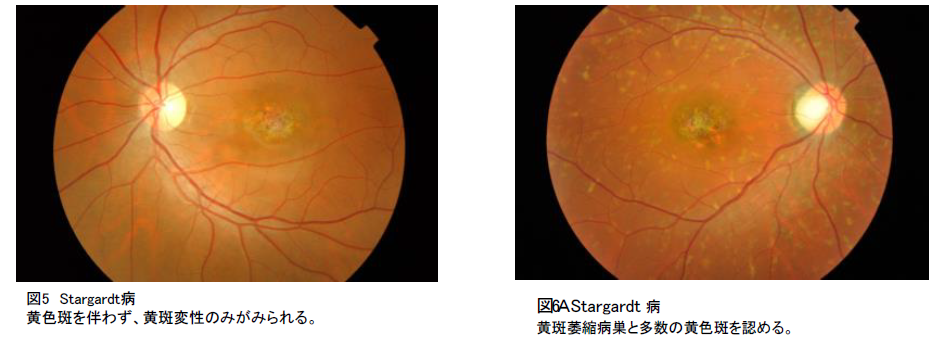
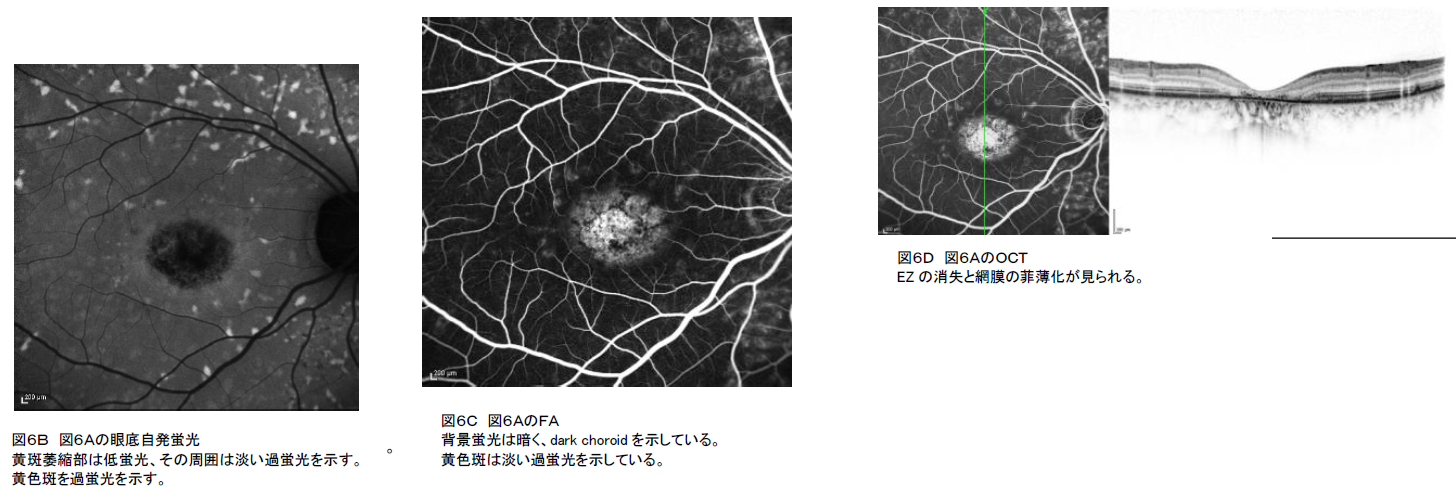
B-① 眼底写真(必須):黄斑の萎縮病巣(典型的萎縮病巣は正常の中心窩領域を取り囲んで輪状の脱色素帯、いわゆる標的黄斑病巣を示すもの(図5)、金属様反射を有する萎縮病巣(beaten-metal atrophy))。典型的な症例では、黄斑周囲あるいは後極部に黄色斑を伴う(図6A)。
B-② 眼底自発蛍光:背景蛍光全体が増強する。黄斑の萎縮病巣は低蛍光、黄色斑は過蛍光を示す(図6B)。また、perpapillary sparing(視神経乳頭周囲の網膜及び色素上皮が温存される所見)も診断に有用である。
蛍光眼底造影(FA 必須):dark choroid(背景蛍光が暗くみえる)が見られる(図6C)。
B-③ 電気生理学的検討:全視野ERG、EOGはさまざまである。黄斑部のERGでは反応減弱が見られる。
B-④ OCT:黄斑部はエリプソイドゾーン(Ellipsoid zone:EZ)の消失と網膜の菲薄化が見られる。黄色斑は網膜色素上皮の瘤状隆起を示す(図6D)。
●診断の要件
診断はB-①眼底写真とB-②蛍光眼底造影、眼底自発蛍光で双方ともに上記の特徴を満たす場合に診断する。
3.オカルト黄斑ジストロフィー
B-① 眼底写真(必須):黄斑部に視力低下を説明できる検眼鏡的な異常がない。
B-② 蛍光眼底造影(FA):黄斑部に視力低下を説明できる異常がない*1。
B-③ 電気生理学的検討(必須):錐体と杆体を分離した全視野網膜電図は正常*2。黄斑局所的ERGの反応が減弱、又は多局所ERGで中心部の反応が減弱。
B-④ OCT:インターデジテーションゾーン(Interdigitation zone:IZ)の消失、EZの不明瞭化が見られる。進行するとEZラインの分断が見られるようになり、外顆粒層も菲薄化する。
●診断の要件
診断はB-①眼底写真、B-②の蛍光眼底造影、及びB-③電気生理学的検討のうち全ての特徴を満たす場合に診断する。
*1 蛍光眼底造影がアレルギーなどで施行困難な場合、眼底自発蛍光で代用することも可能である。眼底自発蛍光は、正常か、もしくは中心窩にわずかな過蛍光が見られる程度の、ごく軽度の異常である。
*2 錐体応答や30-HzフリッカERGは軽度低下することもある。
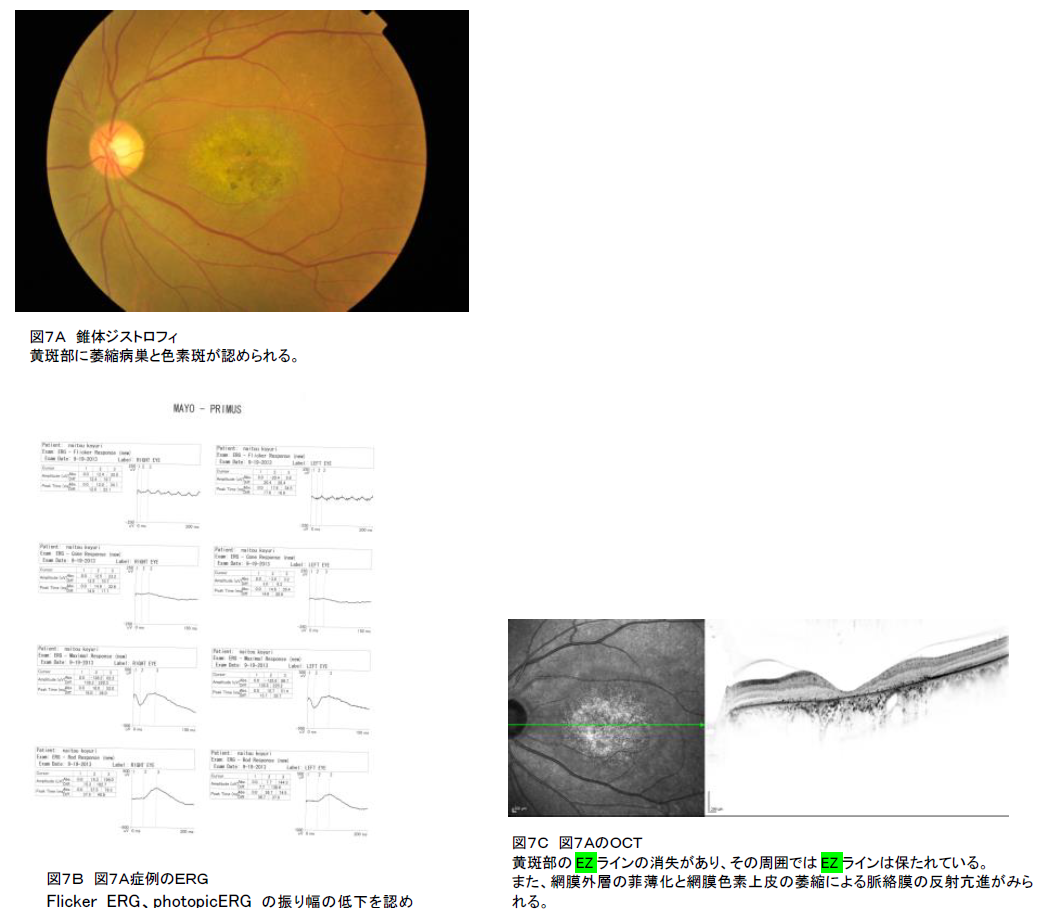
4.錐体ジストロフィー、及び錐体杆体ジストロフィー
B-① 眼底写真(必須):ほとんど異常がないもの、黄斑部に萎縮病巣(典型病巣は標的黄斑病巣、網膜色素上皮のびまん性萎縮(色素沈着を伴うことあり)(図7A))などさまざまである。
B-② 蛍光眼底造影(FA)、眼底自発蛍光(AF):FAでは萎縮に一致してwindow defectによる過蛍光、脈絡毛細血管板萎縮による低蛍光など。AFでは萎縮部位に一致して低蛍光が見られる。病変の境界部に輪状過蛍光が見られることがある。
B-③ 電気生理学的検討(必須):ERGで錐体系ERGの反応減弱(図7B)。杆体系ERGの振幅低下が見られることがある(錐体-杆体ジストロフィー)が、錐体系ERGの異常のほうが高度である。
B-④ OCT: IZは消失する。EZの反射は減弱する。網膜外層の菲薄化が見られる(図7C)。
●診断の要件
診断はB-①眼底写真あるいはB-③(電気生理学的検討)ERGを必須とし、①~④のうち3つ以上の特徴を満たす場合に診断する。
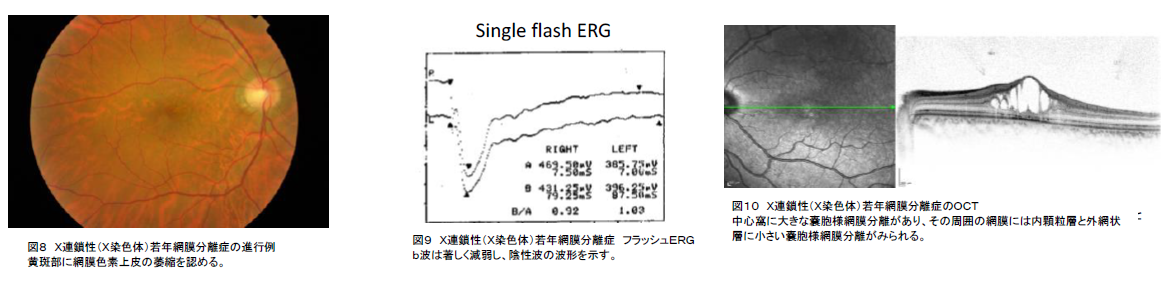
5.X連鎖性(X染色体)若年網膜分離症
(視力:屈折値は遠視であることが多く、眼底変化が目立たない症例では屈折性弱視と誤診されることもある。)
B-① 眼底写真(必須):黄斑に中心窩分離を呈する。進行例では網膜色素上皮の萎縮を伴う非定型的な変性病巣になる(図8)。約半数では周辺部網膜に網膜分離症や網膜反射の異常などを伴う。
B-② 眼底自発蛍光:中心窩の嚢胞に一致し、過蛍光が見られる。
蛍光眼底造影(FA):黄斑分離は蛍光漏出を示さない。
B-③ 電気生理学的検討(必須):フラッシュERGではb波は著しく減弱し、一般にnegative typeを示す(図9)が、若年例では正常型を示すこともある。
B-④ OCT(必須):中心窩周囲の網膜内層、多くは内顆粒層と外網状層に網膜分離所見が認められる(図10)。
●診断の要件
診断はB-①眼底写真あるいはB-④OCTとB-③電気生理学的検討のERGでいずれも上記の特徴を満たす場合に診断する。男性のみに発症することも診断の一助になる。
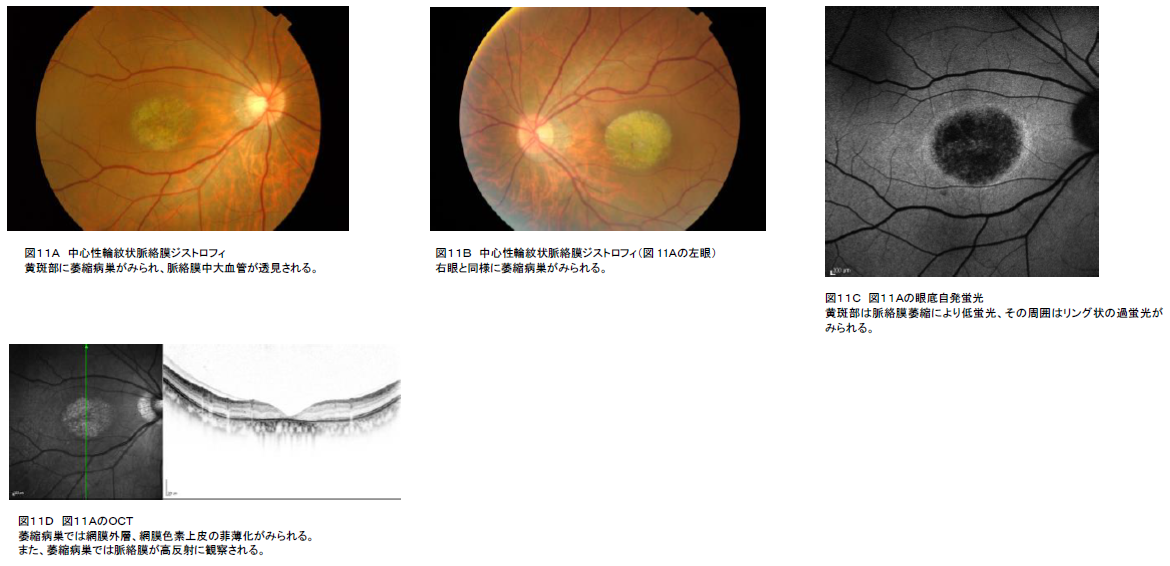
6.中心性輪紋状脈絡膜ジストロフィー
B-① 眼底写真(必須):地図状萎縮病巣内には脈絡膜中大血管が透見される典型病巣を認める。
初期には黄斑あるいは傍黄斑に顆粒状に網膜色素上皮の萎縮病巣が出現する。進行すると網膜色素上皮萎縮病巣内に地図状萎縮病巣が出現し、拡大し、やがて地図状萎縮病巣内には脈絡膜中大血管が透見される典型病巣になる(図11A、図11B)。
B-② 眼底自発蛍光(必須):黄斑部は脈絡膜萎縮により境界鮮明な低蛍光、その辺縁にはリング状の過蛍光が見られる(図11C)。
蛍光眼底造影(FA):初期には病変に一致してwindow defect、進行期には境界鮮明な低蛍光の中に脈絡膜中大血管像が見られる。
B-③ 電気生理学的検討:ERG、EOGは多くの場合正常である。
B-④ OCT:網膜外層、網膜色素上皮の菲薄化が見られる(図11D)。
●診断の要件
診断はB-①眼底写真とB-②蛍光眼底造影で双方ともに上記の特徴を満たす場合に診断する。
<重症度分類>
良好な方の眼の矯正視力が0.3未満の者を対象とする。
※診断基準及び重症度分類の適応における留意事項
1.病名診断に用いる臨床症状、検査所見等に関して、診断基準上に特段の規定がない場合には、いずれの時期のものを用いても差し支えない(ただし、当該疾病の経過を示す臨床症状等であって、確認可能なものに限る。)。
2.治療開始後における重症度分類については、適切な医学的管理の下で治療が行われている状態であって、直近6か月間で最も悪い状態を医師が判断することとする。
3.なお、症状の程度が上記の重症度分類等で一定以上に該当しない者であるが、高額な医療を継続することが必要なものについては、医療費助成の対象とする。
- 黄斑ジストロフィの診断ガイドライン
https://www.nichigan.or.jp/member/journal/guideline/detail.html?itemid=313&dispmid=909 -
<黄斑ジストロフィの疫学>
- Ueno S, Hayashi T, Tsunoda K, Aoki T, Kondo M: Nationwide epidemiologic survey on incidence of macular dystrophy in Japan. Jpn J Ophthalmol: 68(3):167-173, 2024
-
<Stargardt病>
- Kaplan J, Gerber S, Larget-Piet D, et al : A gene for Stargardt’s disease (fundus flavimaculatus) maps to the short arm of chromosome 1. Nat Genet 5 : 308-311, 1993
- Allikmets R : A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet 15 : 236-246, 1997
- Rotenstreich Y, Fishman GA, Anderson RJ : Visual acuity loss and clinical observations in a large series of patients with Stargardt disease. Ophthalmology 110 : 1151-1158, 2003
- Fishman GA, Farber M, Patel BS, Derlacki DJ : Visual acuity loss in patients with Stargardt’s macular dystrophy. Ophthalmology 94 : 809-814, 1987
- Lois N, Holder GE, Bunce C, et al : Phenotypic subtypes of Stargardt macular dystrophy-fundus flavimaculatus. Arch Ophthalmol 119 : 359-369, 2001
- Lois N, Halfyard AS, Bird AC, et al : Fundus autofluorescence in Stargardt macular dystrophy-fundus flavimaculatus. Am J Ophthalmol 138 : 55-63, 2004
- Fujinami K, Lois N, Davidson AE, et al. A longitudinal study of stargardt disease: clinical and electrophysiologic assessment, progression, and genotype correlations. Am J Ophthalmol. 2013;155(6):1075-1088 e1013.
- Mizobuchi K, Hayashi T, Tanaka K,et al: Genetic and Clinical Features of ABCA4-Associated Retinopathy in a Japanese Nationwide Cohort.Am J Ophthalmol; 264: 36-43, 2024
-
<錐体杆体ジストロフィー>
- Ito S, Nakamura M, Ohnishi Y, Miyake Y : Autosomal dominant cone-rod dystrophy with R838H and R838C mutations in the GUCY2D gene in Japanese patients. Jpn J Ophthalmol 48 : 228-235, 2004
- Paunescu K, Preising MN, Janke B, et al : Genotype-phenotype correlation in a German family with a novel complex CRX mutation extending the open reading frame. Ophthalmology 114 : 1348-1357, 2007
- Michaelides M, Holder GE, Hunt DM, et al : A detailed study of the phenotype of an autosomal dominant cone-rod dystrophy (CORD7) associated with mutation in the gene for RIM1. Br J Ophthalmol 89 : 198-206, 2005
- Hameed A, Abid A, Aziz A, et al : Evidence of RPGRIP1 gene mutations associated with recessive cone-rod dystrophy. J Med Genet 40 : 616-619, 2003
- Fujinami K, Tsunoda K, Nakamura N, et al. Molecular characteristics of four Japanese cases with KCNV2 retinopathy: report of novel disease-causing variants. Molecular vision. 2013;19:1580-1590.
- Mears, A. J., Hiriyanna, S., Vervoort, R., Yashar, B., Gieser, L., Fahrner, S., Daiger, S. P., Heckenlively, J. R., Sieving, P. A., Wright, A. F., Swaroop, A. Remapping of the RP15 locus for X-linked cone-rod degeneration to Xp11.4-p21.1, and identification of a de novo insertion in the RPGR exon ORF15. Am. J. Hum. Genet. 67: 1000-1003, 2000.
-
<卵黄状黄斑ジストロフィー>
- Ito S, Nakamura M, Ohnishi Y, Miyake Y : Autosomal dominant cone-rod dystrophy with R838H and R838C mutations in the GUCY2D gene in Japanese patients. Jpn J Ophthalmol 48 : 228-235, 2004
- Paunescu K, Preising MN, Janke B, et al : Genotype-phenotype correlation in a German family with a novel complex CRX mutation extending the open reading frame. Ophthalmology 114 : 1348-1357, 2007
- Michaelides M, Holder GE, Hunt DM, et al : A detailed study of the phenotype of an autosomal dominant cone-rod dystrophy (CORD7) associated with mutation in the gene for RIM1. Br J Ophthalmol 89 : 198-206, 2005
- Hameed A, Abid A, Aziz A, et al : Evidence of RPGRIP1 gene mutations associated with recessive cone-rod dystrophy. J Med Genet 40 : 616-619, 2003
- Fujinami K, Tsunoda K, Nakamura N, et al. Molecular characteristics of four Japanese cases with KCNV2 retinopathy: report of novel disease-causing variants. Molecular vision. 2013;19:1580-1590.
- Mears, A. J., Hiriyanna, S., Vervoort, R., Yashar, B., Gieser, L., Fahrner, S., Daiger, S. P., Heckenlively, J. R., Sieving, P. A., Wright, A. F., Swaroop, A. Remapping of the RP15 locus for X-linked cone-rod degeneration to Xp11.4-p21.1, and identification of a de novo insertion in the RPGR exon ORF15. Am. J. Hum. Genet. 67: 1000-1003, 2000.
-
<X連鎖性若年網膜分離症>
- Sauer CG, Gehrig A, Warneke-Wittstock R, et al : Positional cloning of the gene associated with X-linked juvenile retinoschisis. Nat Genet 17 : 164-170, 1997
- George ND, Yates JR, Moore AT : X linked retinoschisis. Br J Ophthalmol 79 : 697-702, 1995
- George ND, Yates JR, Moore AT : Clinical features in affected males with X-linked retinoschisis. Arch Ophthalmol 114 : 274-280, 1996
- Hotta Y, Fujiki K, Hayakawa M, et al. Japanese juvenile retinoschisis is caused by mutations of the XLRS1 gene. Human genetics. 1998;103(2):142-144.
- Shinoda K, Ohde H, Mashima Y, et al : On- and off-responses of the photopic electroretinograms in X-linked juvenile retinoschisis. Am J Ophthalmol 131 : 489-494, 2001
- Hayashi, T., Omoto, S., Takeuchi, T., Kozaki, K., Ueoka, Y., Kitahara, K. Four Japanese male patients with juvenile retinoschisis: only three have mutations in the RS1 gene. Am. J. Ophthal. 138: 788-798, 2004.
-
<オカルト黄斑ジストロフィー>
- Miyake Y, Ichikawa K, Shiose Y, Kawase Y. Hereditary macular dystrophy without visible fundus abnormality. Am J Ophthalmol. 1989;108(3):292-299.
- Miyake Y, Horiguchi M, Tomita N, et al. Occult macular dystrophy. Am J Ophthalmol. 1996;122(5):644-653.
- Akahori M, Tsunoda K, Miyake Y, et al. Dominant Mutations in RP1L1 Are Responsible for Occult Macular Dystrophy. Am J Hum Genet. 2010;87(3):424-429.
- Tsunoda K, Usui T, Hatase T, et al. Clinical Characteristics of Occult Macular Dystrophy in Family with Mutation of Rp1l1 Gene. Retina-the Journal of Retinal and Vitreous Diseases. 2012;32(6):1135-1147.
-
<中心性輪紋状網脈絡膜ジストロフィー>
- Wells, J., Wroblewski, J., Keen, J., Inglehearn, C., Jubb, C., Eckstein, A., Jay, M., Arden, G., Bhattacharya, S., Fitzke, F., Bird, A. Mutations in the human retinal degeneration slow (RDS) gene can cause either retinitis pigmentosa or macular dystrophy. Nature Genet. Mar;3(3):213-8, 1993.
- Hughes AE, Meng W, Lotery AJ, Bradley DT. A novel GUCY2D mutation, V933A, causes central areolar choroidal dystrophy. Invest Ophthalmol Vis Sci. 2012;53(8):4748-4753.
-
<日本語総説>
- 三宅養三「黄斑ジストロフィー」日本眼科学会雑誌 107巻4号2003; 229-241
- 近藤峰生「黄斑ジストロフィーの診断」日本の眼科 83:4号2012; 456-463
- 藤波芳、角田和繁「黄斑ジストロフィーの遺伝子異常」眼科 Vol.53, No.2, 2011; 239-255
- 角田和繁、藤波芳「黄斑ジストロフィー(三宅病を含めて)」眼科 Vol. 56, No. 5, 2014; 575-584
- 角田和繁「卵黄様黄斑ジストロフィー」眼科 Vol. 57, No. 4, 2015; 641-645
治験を実施している指定難病を調べることができます。キーワード検索欄に指定難病名を入力し、検索してください。
治験・臨床研究情報検索サイト一覧
| 研究班名 | 網膜脈絡膜・視神経萎縮症に関する調査研究班 研究班名簿 |
|---|---|
| 情報更新日 | 令和6年10月(名簿更新:令和8年6月) |