無βリポタンパク血症(指定難病264)
1. 「無βリポタンパク血症」とはどのような病気ですか
コレステロールや中性脂肪などの脂質は、食事から取り込まれたり、体の中で合成されたりして、最終的には血中を運ばれて全身の組織で利用されています。このような脂質は水に溶けないため,血液の中では「リポタンパク」という粒子で存在します。無βリポタンパク血症は、リポタンパクを作るMTTPという遺伝子に病的バリアント(病気に関わる遺伝子の変化)があるために、脂質が十分に吸収・運搬できなくなり、様々な合併症を引き起こしてしまう病気です(図)。
食事で摂取した脂質は小腸で吸収され、体の各組織に運ばれます。しかし、この病気では、小腸でリポタンパクが作られないために、脂質の吸収ができなくなり、下痢や成長障害をきたし、血液中のコレステロール値や中性脂肪値が非常に低くなります(低脂血症といいます)。脂肪の吸収は、脂溶性ビタミンであるビタミンE、A、D、Kの吸収にも関わっているため、脂溶性ビタミンの欠乏による様々な症状(眼症状、神経症状など)が起こります。
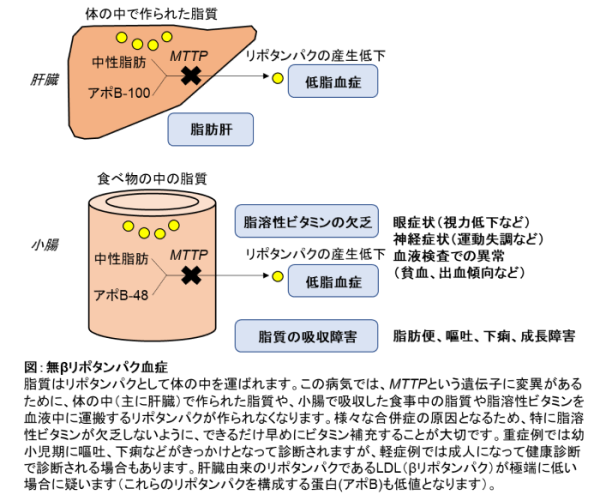
体の中で合成された脂質は主に肝臓でリポタンパクとなり、各組織に運ばれます。しかし、この病気では、MTTP遺伝子の病的バリアントが原因で肝臓でリポタンパクが作られないために、肝臓に脂肪がたまり、脂肪肝となります。
肝臓でつくられたリポタンパクは、最終的にLDL(βリポタンパク)と呼ばれるリポタンパクになるのですが、この病気ではLDLがほとんど無くなることから「無βリポタンパク血症」と呼ばれています。無βリポタンパク血症(英語でabetalipoproteinemiaを略してABLと呼ばれます)は、1950年に最初に報告されました。
2. この病気の患者さんはどのくらいいるのですか
無βリポタンパク血症の患者さんは100万人に1人以下と言われています。世界で約100例の報告があります。日本では約10家系程度の報告があります。
3. この病気はどのような人に多いのですか
体の中の遺伝子は、通常、父親由来と母親由来の2つの遺伝子を受け継いでいます。無βリポタンパク血症の場合には、通常、父親由来と母親由来のMTTP遺伝子の両方に病的バリアント(病気に関わる遺伝子の変化)がある場合(ホモ接合体といいます)に病気となります。したがって、ご両親に血族結婚がある場合には、病的バリアントが集積しやすく、発症する可能性が高くなります。ご両親に血族結婚がない場合でも、ご両親が同じ地域(血族結婚の多い地域、人の移動の少ない地域)のご出身の場合にも、このような病的バリアントが重なることがあります。
出生時までは明らかな症状は見られないものの、重症例では、授乳開始とともに脂肪吸収障害による脂肪便、慢性下痢、嘔吐が出現して気付かれます。他に、視力低下などの眼症状、運動失調などの神経症状が、個人差はありますが徐々に現れてきます。
軽症例では、成人になってから、血液検査でコレステロールやトリグリセリドなどが非常に低いことが診断のきっかけとなる場合もあります。
4. この病気の原因はわかっているのですか
この病気の原因は、小腸で吸収された脂肪や、肝臓で合成された脂肪を血中に運搬する粒子(リポタンパク)をつくるのに必要な「ミクロソームトリグリセリド転送蛋白(microsomal triglyceride transfer protein(MTTP))」の遺伝子に病的バリアントがあることです。
5. この病気は遺伝するのですか
ヒトの体は、基本的に、父親由来と母親由来の2つの遺伝子を受け継いでいます。MTTP遺伝子の場合も、両親から1つずつ、合わせて2つ受け継ぎます。この病気の場合、MTTP遺伝子1つだけに病的バリアントがある場合(ヘテロ接合体といいます)には発症せず、2つとも病的バリアントがある場合(ホモ接合体といいます)に発症します。つまり無βリポタンパク血症の患者さんはご両親から、MTTP遺伝子の病的バリアントを1つずつ受け継いでいることになります。ご両親は、MTTP遺伝子の1つだけに病的バリアントがあるヘテロ接合体のため、病気は発症しません(ただし病気の原因となる遺伝子は持っており、「保因者」と呼ばれます)。無βリポタンパク血症の患者さんのご兄弟(姉妹)は、1/4の確率で同じ病気(ホモ接合体)である可能性があり、1/2の確率で保因者(ヘテロ接合体)である可能性があります。無βリポタンパク血症の患者さんと病的バリアントの無い方から生まれたお子さんは保因者(ヘテロ接合体)になりますので、重篤な病気を発症することはありません。
6. この病気ではどのような症状がおきますか
授乳開始時より、脂肪吸収障害による脂肪便、慢性下痢、嘔吐、腹部膨満が出現し、発育障害をきたすことが多くなります。また、脂溶性ビタミン(ビタミンE、A、D、K)の吸収障害に伴って、個人差はありますが、思春期までに神経症状や眼症状など様々な症状が現れてきます。主にビタミンE欠乏によって神経症状(脊髄小脳変性による運動失調、末梢神経障害による知覚低下など)が生じ、主にビタミンEやビタミンAの欠乏により眼症状(夜盲症、色覚異常、網膜色素変性症、視力低下など)が生じます。他にも、ビタミンD欠乏によるくる病や骨軟化症といった骨の成長障害や、ビタミンK欠乏による出血傾向や不整脈を伴う心臓病が出現することもあるとされています。しばしば脂肪肝を認め、肝硬変や肝臓がんの合併の報告もあります。棘の生えたような形に赤血球が変形することもあります(このような赤血球は「有棘赤血球」と呼ばれます)。
7. この病気にはどのような治療法がありますか
脂溶性ビタミンの補充療法が行われます。出来るだけ早期からの投与が重要です。投与は経口の内服で行いますが、腸管からの吸収が障害されていますので大量に投与する必要があります。網膜色素変性や神経変性の進行を抑えるためには、特にビタミンEやビタミンAの補充が重要とされています。下痢、嘔吐などの消化器症状については脂肪制限が行われます。吸収できる脂質として、中鎖トリグリセリド(medium chain triglyceride(MCT))が用いられることもあります。ビタミンDやビタミンK不足がある場合にはそれらの補充を行います。
8. この病気はどういう経過をたどるのですか
経過には個人差がありますが、典型例は成人までに網膜色素変性による視力障害および脊髄小脳変性による運動失調、末梢神経障害による知覚低下など多彩な神経症状が出現し、失明や歩行障害などが徐々に進行します。脂肪肝を合併することが多く、肝硬変や肝臓がんに進展したという報告もあります。幼児期から脂溶性ビタミンを大量投与することによって、これらの視力障害や神経障害を抑えることが出来たという報告があり、60~70歳代までわずかな症状のみで推移されている方も報告されています。眼の症状や神経の症状の評価のために、定期的な眼科受診や神経の診察が必要になります。
9. この病気は日常生活でどのような注意が必要ですか
合併症の進行を防ぐため、治療を中断せずに、脂溶性ビタミンの補充をしっかりと続けることが大切です。脂肪吸収障害による下痢などの消化器症状を軽減するためには、脂肪摂取(特に長鎖脂肪酸)を制限することが推奨されます(脂肪摂取量は1日の総カロリーの30%以下が目安となります)。脂肪吸収障害は、それに引き続いて炭水化物やタンパク質などのその他の栄養素の吸収も障害してしまう(これを続発的吸収障害といいます)ため、脂肪摂取制限は栄養バランスの改善にも役立つとされています。低栄養とならないように、適切なカロリーを摂取するようにします。吸収できる脂質として、中鎖トリグリセリド(MCT)を用いることもあります。また、体にとって必要な脂肪酸(必須脂肪酸)が不足しないように、許容範囲内で1日小さじ1杯程度の大豆油やオリーブオイルのような多価不飽和脂肪酸の摂取も考慮します。妊娠を希望する場合は、ビタミンAの過剰が胎児に先天異常を来す可能性があるため、補充量の調整のために、事前に医師に相談する必要があります。ビタミンAは必須のビタミンであることから、妊娠中もビタミンA補充を中止せず、継続する必要があります。脊髄小脳変性による運動失調がみられるようになった場合には、身体機能維持のためのリハビリテーションや、転倒防止のための杖や歩行器の使用を考慮します。
10. 次の病名はこの病気の別名又はこの病気に含まれる、あるいは深く関連する病名です。 ただし、これらの病気(病名)であっても医療費助成の対象とならないこともありますので、主治医に相談してください。
深く関連する疾患:家族性低βリポタンパク血症(FHBL)1(ホモ接合体)(指定難病336)
別名:MTP欠損症
治験を実施している指定難病を調べることができます。キーワード検索欄に指定難病名を入力し、検索してください。
治験・臨床研究情報検索サイト一覧