原発性免疫不全症候群(指定難病65)
○ 概要
1.概要
原発性免疫不全症候群は、先天的に免疫系のいずれかの部分に欠陥がある疾患の総称であり、後天的に免疫力が低下するエイズなどの後天性免疫不全症候群と区別される。障害される免疫担当細胞(例えば、好中球、T細胞、B細胞)などの種類や部位により200近くの疾患に分類される。
原発性免疫不全症候群で問題となるのは、感染に対する抵抗力の低下である。重症感染のため重篤な肺炎、中耳炎、膿瘍、髄膜炎などを繰り返す。時に生命の危険を生じることもあり、中耳炎の反復による難聴、肺感染の反復により気管支拡張症などの後遺症を残すこともある。
2.原因
多くは免疫系に働く蛋白の遺伝子の異常である。この10年間に代表的な原発性免疫不全症候群の原因遺伝子は多くが解明され、確定診断や治療に役立っている。しかし、IgGサブクラス欠乏症の一部、乳児一過性低γグロブリン血症のように一時的な免疫系の未熟性、慢性良性好中球減少症のように自己抗体によると思われる疾患もある。
3.症状
主な症状は易感染性である。つまり、風邪症状がなかなか直らなかったり、何度も発熱したりし、入院治療が必要である。重症のタイプでは感染が改善せず、致死的となることもある。好中球や抗体産生の異常による疾患では細菌感染が多く、T細胞などの異常ではウイルスや真菌感染が多い傾向がある。
原発性免疫不全症を疑う10の徴候があり、以下に示す。
1.乳児で呼吸器・消化器感染症を繰り返し、体重増加不良や発育不良が見られる。
2.1年に2回以上肺炎にかかる。
3.気管支拡張症を発症する。
4.2回以上、髄膜炎、骨髄炎、蜂窩織炎、敗血症や、皮下膿痬、臓器内膿痬などの深部感染症にかかる。
5.抗菌薬を服用しても2か月以上感染症が治癒しない。
6.重症副鼻腔炎を繰り返す。
7.1年に4回以上、中耳炎にかかる。
8.1歳以降に、持続性の鵞口瘡、皮膚真菌症、重度・広範な疣贅(いぼ)が見られる。
9.BCGによる重症副反応(骨髄炎など)、単純ヘルペスウイルスによる脳炎、髄膜炎菌による髄膜炎、EBウイルスによる重症血球貧食症候群に罹患したことがある。
10.家族が乳幼児期に感染症で死亡するなど、原発性免疫不全症候群を疑う家族歴がある。
これらの所見のうち1つ以上当てはまる場合は、原発性免疫不全症の可能性がないか専門の医師に相談する。この中で、乳児期早期に発症することの多い重症複合免疫不全症は緊急に治療が必要である。
4.治療法
疾患・重症度により治療法が選択される。
軽症例では、抗菌薬、抗ウイルス剤、抗真菌剤の予防内服が効果的である。抗体欠乏を主徴とする免疫不全症では、月1回ほどの静注用ヒト免疫グロブリン製剤の補充により感染はほぼ予防できる。好中球減少症ではG-CSFの定期投与、慢性肉芽腫症ではIFN-γの定期投与が効果ある。
重症複合免疫不全症などの重症なタイプでは、早期に骨髄や臍帯血による造血幹細胞移植が選択される。ドナーが見つからない場合は遺伝子治療が考慮される。
5.予後
疾患や重症度によりかなり異なる。軽症例では、抗菌薬の予防内服やヒト免疫グロブリンの補充療法などにより、通常の日常生活が送れる。それに対し、重症複合免疫不全症などは、造血幹細胞移植をしないと多くは2歳以上まで生存できない。また、慢性肉芽腫症などは予防内服をしていても、30歳以上になるとかなり予後不良となる。なによりも、まれな疾患でもあり専門の施設での診断、治療、経過観察が大切である。
○ 要件の判定に必要な事項
1.患者数(平成24年度医療受給者証保持者数)
1,383人
2.発病の機構
不明(遺伝子の異常)
3.効果的な治療方法
未確立(対症療法のみで根治的療法なし。)
4.長期の療養
必要(継続的な感染症対策が必要。)
5.診断基準
あり(現行の特定疾患治療研究事業の診断基準を研究班にて改訂)
6.重症度分類
研究班による重症度分類を用いて中等症以上を対象とする。
○ 情報提供元
「原発性免疫不全症候群の診断基準・重症度分類および診療ガイドラインの確立に関する研究班」
研究代表者 防衛医科大学校小児科学 教授 野々山恵章
<診断基準>
国際免疫学会の原発性免疫不全症分類専門委員による分類に準じ、原発性免疫不全症候群の診断基準・重症度分類および診療ガイドラインの確立に関する研究班及び日本免疫不全症研究会の作製した診断基準を用いる。
1.主要項目
(1) 原発性免疫不全症候群に含まれる疾患 (国際免疫学会の分類に準ずる。)
① 複合免疫不全症
Ⅰ.X連鎖重症複合免疫不全症
Ⅱ.細網異形成症
Ⅲ.アデノシンデアミナーゼ(ADA)欠損症
Ⅳ.オーメン(Omenn)症候群
Ⅴ.プリンヌクレオシドホスホリラーゼ欠損症
Ⅵ.CD8欠損症
Ⅶ.ZAP-70欠損症
Ⅷ.MHCクラスI欠損症
Ⅸ.MHCクラスII欠損症
Ⅹ.IからXまでに掲げるもののほか、複合免疫不全症
② 免疫不全を伴う特徴的な症候群
Ⅰ.ウィスコット・オルドリッチ(Wiskott-Aldrich)症候群
Ⅱ.毛細血管拡張性運動失調症
Ⅲ.ナイミーヘン染色体不安定(Nijmegen breakage)症候群
Ⅳ.ブルーム(Bloom)症候群
Ⅴ.ICF症候群
Ⅵ.PMS2異常症
Ⅶ.RIDDLE症候群
Ⅷ.シムケ(Schimke)症候群
Ⅸ.ネザートン(Netherton)症候群
Ⅹ.胸腺低形成(DiGeorge)症候群、 22q11.2欠失症候群)
Ⅺ.高IgE症候群
Ⅻ.肝中心静脈閉鎖症を伴う免疫不全症
XⅢ.先天性角化不全症
③ 液性免疫不全を主とする疾患
Ⅰ.X連鎖無ガンマグロブリン血症
Ⅱ.分類不能型免疫不全症
Ⅲ.高IgM症候群
Ⅳ.IgGサブクラス欠損症
Ⅴ.選択的IgA欠損症
Ⅵ.特異抗体産生不全症
Ⅶ.乳児一過性低ガンマグロブリン血症
Ⅷ.IからVIIまでに掲げるものの他、液性免疫不全を主とする疾患
④ 免疫調節障害
Ⅰ.チェディアック・東(Chédiak-Higashi)症候群
Ⅱ.X連鎖リンパ増殖症候群
Ⅲ.自己免疫性リンパ増殖症候群(ALPS)
Ⅳ.IからIIIに掲げるもののほか、免疫調節障害
⑤ 原発性食細胞機能不全症及び欠損症
Ⅰ.症先天性好中球減少症
Ⅱ.周期性好中球減少症
Ⅲ.IおよびIIに掲げるもののほか、慢性の経過をたどる好中球減少症
Ⅳ.白血球接着不全症
Ⅴ.シュワッハマン・ダイアモンド(Shwachman-Diamond)症候群
Ⅵ.慢性肉芽腫症
Ⅶ.ミエロペルオキシダーゼ欠損症
Ⅷ.メンデル遺伝型マイコバクテリア易感染症
Ⅸ.IVからVIIIに掲げるもののほか、白血球機能異常
⑥ 自然免疫異常
Ⅰ.免疫不全を伴う無汗性外胚葉形成異常症
Ⅱ.IRAK4欠損症
Ⅲ.MyD88欠損症
Ⅳ.慢性皮膚粘膜カンジダ症
Ⅴ.IからIVに掲げるもののほか、自然免疫異常
⑦ 先天性補体欠損症
Ⅰ.先天性補体欠損症
Ⅱ.遺伝性血管性浮腫(C1インヒビター欠損症)
Ⅲ.I及びIIに掲げるもののほか、先天性補体欠損症
(2) 除外事項
続発性免疫不全状態を来すことの多い慢性代謝性疾患、染色体異常、HIVなどのウイルス感染、 悪性腫瘍や抗癌剤、免疫抑制剤投与、移植などによる医原性免疫不全状態が除外されていること。
2.参考事項
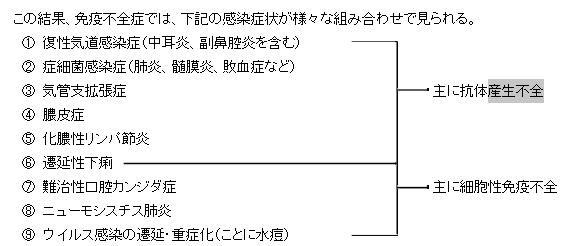
免疫不全症の多くに共通して見られる易感染性は、次のように要約される。
(1) 様々な部位の頻回の罹患傾向に加え、個々の感染が重症化しやすく、治癒が遷延する。
(2) 肺炎、髄膜炎、敗血症など重症感染症の反復罹患
(3) ニューモシスチス、カンジダ、サイトメガロウイルスなどの日和見感染
<診断基準>
(1) 原発性免疫不全症候群に含まれる疾患(国際免疫学会の分類に準ずる。)
①複合免疫不全症
<I.X連鎖重症複合免疫不全症>
1.通常生後数か月以内に日和見感染を含む様々な重症感染症を発症し、根治的治療である造血幹細胞移植を行わなければ生後1年以内に死亡する。
2.基本的には男児に発症
3.通常末梢血T細胞とNK細胞数は欠損又は著減し(<300/µL)、B細胞数は正常(T-B+NK-)。
4.PHA幼若化反応が正常の10%未満
5.無~低ガンマグロブリン血症:出生後数か月間は母体からのIgG型移行抗体が存在するため必ずしも低値とならない。また、IgG値の正常値は月齢や年齢によって大きく異なる。
6.common γ(γc)鎖遺伝子の異常による。
γc遺伝子解析で遺伝子異常を確認し、確定診断を行う。Primary Immunodeficiency Database in Japan(PIDJ)(https://jsiad.org/consultation/)の患者相談フォームで相談することが可能、さらに、一部の施設ではフローサイトメトリー法でリンパ球表面γc鎖発現解析も行っている。
まれに母からのT細胞が生着したり、変異が一部のリンパ球分画で正常に戻る(reversion)現象が観察されており、T細胞が存在する例も存在するので、専門施設に早期に相談することが望ましい。
<II.細網異形成症>
1.通常生後数か月以内に日和見感染を含む様々な重症感染症を発症し、根治的治療である造血幹細胞移植を行わなければ生後1年以内に死亡する。
2.男児、女児いずれにも発症する。
3.末梢血T細胞は欠損又は著減:<300/µLし、
好中球も欠損又は著減:<200/µL
4.典型例では感音性難聴を呈する。
5.PHA幼若化反応が正常の10%未満
6.骨髄系細胞分化障害の骨髄所見
7.無~低ガンマグロブリン血症:出生後数か月間は母体からのIgG型移行抗体が存在するため必ずしも低値とならない。また、IgG値の正常値は月齢や年齢によって大きく異なる。
8.非典型例では再生不良性貧血、骨髄異形成症候群、骨髄不全との鑑別が困難である。
9.adenylate kinase 2(AK2)遺伝子の異常による。
AK2遺伝子解析で遺伝子異常を確認し、確定診断を行う。
Primary Immunodeficiency Database in Japan(PIDJ)(https://jsiad.org/consultation/)の患者相談フォームで相談することが可能
<III.アデノシンデアミナーゼ(ADA)欠損症>
1.通常生後数か月以内に日和見感染を含む様々な重症感染症を発症し、根治的治療である造血幹細胞移植を行わなければ生後1年以内に死亡する。
2.男児、女児いずれにも発症する。
3.通常末梢血リンパ球が全て欠損又は著減(<500/µL)し(T-B-NK-)、T細胞は欠損または著減:<300/µL
4.PHA幼若化反応が正常の10%未満
5.無~低ガンマグロブリン血症:出生後数か月間は母体からのIgG型移行抗体が存在するため必ずしも低値とならない。また、IgG値の正常値は月齢や年齢によって大きく異なる。
6.発達遅滞、痙攣、難聴の合併などが見られる。
7.末梢血単核球、赤血球、線維芽細胞などのADA活性が低下
8.ADA遺伝子の異常による。
ADA遺伝子解析で遺伝子異常を確認し、確定診断を行う。
Primary Immunodeficiency Database in Japan(PIDJ)(https://jsiad.org/consultation/)の患者相談フォームで相談することが可能。さらに、北海道大学小児科では末梢血単核球、赤血球や線維芽細胞のADA活性測定が可能。まれに母からのT細胞が生着したり、変異が一部のリンパ球分画で正常に戻る(reversion)現象が観察されており、リンパ球が存在する例も存在するので、専門施設に早期に相談することが望ましい。
<IV.オーメン(Omenn)症候群>
通常生後数か月以内に日和見感染を含む様々な重症感染症を発症し、根治的治療である造血幹細胞移植を行わなければ生後1年以内に死亡する。
1.特徴的臨床症状
生後まもなくよりの湿疹様皮膚病変、リンパ節腫大、肝脾腫、易感染性など
2.特徴的検査所見
末梢血T細胞は存在(>300/µL)し、好酸球増加、高IgE血症を伴う。
3.RAG1、RAG2を含む重症複合免疫不全症の責任遺伝子の異常による。
RAG1、RAG2などの遺伝子解析で遺伝子異常を確認し、確定診断を行う。
Primary Immunodeficiency Database in Japan(PIDJ)(https://jsiad.org/consultation/)の患者相談フォームで相談することが可能
<V.プリンヌクレオシドホスホリラーゼ欠損症>
1.男児、女児いずれにも発症する。
2.血清尿酸値の低下(<1mg/mL)
3.通常末梢血T細胞が進行性に減少し、B細胞数は正常B細胞が減少する場合もある。
4.末梢血単核球、赤血球、線維芽細胞などのPNP活性が低下
5.PNP遺伝子の異常による。
PNP遺伝子解析で遺伝子異常を確認し、確定診断を行う。Primary Immunodeficiency Database in Japan(PIDJ)(https://jsiad.org/consultation/)の患者相談フォームで相談することが可能
<VI.CD8欠損症>
1.男児、女児いずれにも発症する。
2.通常末梢血リンパ球は正常だが、CD8陽性細胞が欠損CD8α遺伝子解析で遺伝子異常を確認し、確定診断を行う。
Primary Immunodeficiency Database in Japan(PIDJ)(https://jsiad.org/consultation/)の患者相談フォームで相談することが可能
<VII.ZAP-70欠損症>
1.男児、女児いずれにも発症する。
2.通常末梢血リンパ球、T細胞は正常だが、CD8陽性細胞は欠損または著減(0~5%)
3.PHA幼若化反応が正常の10%未満
4.ZAP-70遺伝子の異常による。
ZAP-70遺伝子解析で遺伝子異常を確認し、確定診断を行う。Primary Immunodeficiency Database in Japan(PIDJ)(https://jsiad.org/consultation/)の患者相談フォームで相談することが可能
<VIII.MHCクラスI欠損症>
1.男児、女児いずれにも発症するが、無症状の場合もある。
2.CD8陽性細胞が減少
3.リンパ球細胞表面MHC class Iの発現が欠損又は低下
4.NK細胞活性化が低下
5.既知の責任遺伝子はTAP1、TAP2、TAPBP
TAP1、TAP2、TAPBP遺伝子解析で遺伝子異常を確認し、確定診断を行う。
Primary Immunodeficiency Database in Japan(PIDJ)(https://jsiad.org/consultation/)の患者相談フォームで相談することが可能
<IX.MHCクラスII欠損症>
1.男児、女児いずれにも発症する。
2.通常末梢血リンパ球、T細胞数は正常だが、CD4陽性細胞が減少
3.B細胞表面MHC class IIの発現が欠損
4.無~低ガンマグロブリン血症:出生後数か月間は母体からのIgG型移行抗体が存在するため必ずしも低値とならない。またIgG値の正常値は月齢や年齢によって大きく異なる。
5.既知の責任遺伝子はRFXANK、CIITA、RFX5、RFXAP
RFXANK、CIITA、RFX5、RFXAP遺伝子解析で遺伝子異常を確認し、確定診断を行う。
Primary Immunodeficiency Database in Japan(PIDJ)(https://jsiad.org/consultation/)の患者相談フォームで相談することが可能
<IからIXまでに掲げるもののほか、複合免疫不全症>
複合免疫不全症(CID)はT細胞系、B細胞系両者の免疫不全を伴った疾患の総称である。2011年のIUIS分類の段階でも30以上のCID責任遺伝子が明らかになっており、今後も更に増えることが予想される。
2013年に提唱されたCID診断criteria(JACI、 Nov27)によると、重症型CID(SCID)は、末梢血T細胞が欠損又は著減し(<300/µL)、PHA幼若化反応が正常の10%未満のもの、それよりも軽症なCID(leaky SCID)は、末梢血T細胞が2~4歳<800 µL、4歳~<600 µL PHA幼若化反応が正常の30%未満のものと分類されている。多くのCIDは、リンパ球やそれぞれのリンパ球分画の減少の有無などによってある程度鑑別は可能である。
多くのCIDは、リンパ球やそれぞれのリンパ球分画の減少の有無などによって、ある程度鑑別は可能である。しかし、最終的な確定診断のためには遺伝子診断が必要である。CIDの責任遺伝子解析についてはPrimary Immunodeficiency Database in Japan(PIDJ)(https://jsiad.org/consultation/)の患者相談フォームで相談することが可能。
②免疫不全を伴う特徴的な症候群
<I.ウィスコット・オルドリッチ(Wiskott-Aldrich)症候群>
診断方法
A.主要臨床症状
1.易感染性
易感染性の程度は症例により異なるのが特徴である。
2.血小板減少
ほぼ全例で見られ、血便、皮下出血が多い。小型血小板を伴う。
3.湿疹
湿疹はアトピー性湿疹様で、難治である。
B.重要な検査所見
1.小型血小板を伴う血小板減少を伴う。
2.T細胞数の減少とCD3抗体刺激に対する反応低下が見られる。
3.B細胞では免疫グロブリンはIgM低下、IgA上昇、IgE上昇を認める。抗多糖類抗体、同種血球凝集素価などの特異抗体産生は低下する。
4.NK活性は半数で低下する。
5.補体価は正常とされるが、好中球および単球の遊走能は低下する例が多い。
確定診断には、フローサイトメトリー法によるWASP蛋白発現低下とWASPあるいはWIP遺伝子変異を同定する。WASP遺伝子変異はX連鎖性、WIP遺伝子変異は常染色体劣性遺伝形式をとる。
<II.毛細血管拡張性運動失調症>
診断方法:
A.主要臨床症状
1.歩行開始と共に明らかになる歩行失調(体幹失調):必発症状
徐々に確実に進行(2歳から5歳までの間には進行がマスクされることもある。)
2.小脳性構語障害・流涎
3.眼球運動の失行、眼振
4.舞踏病アテトーゼ(全例ではない。)
5.低緊張性顔貌
6.眼球結膜・皮膚の毛細血管拡張
6歳までに50%、8歳時で90%が明らかになる。
7.免疫不全症状(反復性気道感染症)
但し30%では免疫不全症状を認めない。
8.悪性腫瘍:特にT細胞性腫瘍の発生頻度が高い。
9.その他:
発育不良、内分泌異常(耐糖能異常:インスリン非依存性糖尿病)、皮膚、頭髪、血管の早老性変化
B.重要な検査所見
1.αフェトプロテインの上昇(2歳以降、95%で)
2.CEAの増加(認めることがある。)
3.IgG(IgG2)、IgA、IgEの低下
4.T細胞数の低下、CD4陽性T細胞中CD4+CD45RA+細胞の比率の低下
5.電離放射線高感受性
リンパ球と線維芽細胞の染色体不安定性
確定診断には、ATM蛋白発現低下とATM遺伝子変異を同定する。常染色体劣性遺伝形式をとる。
類縁疾患として、Ataxia-telagiectasia like disease(ATLD)があり、MRE11遺伝子異常を伴う。常染色体劣性遺伝形式をとる。
<III.ナイミーヘン染色体不安定(Nijmegen breakage)症候群>
診断方法:
A.主要臨床症状
1.小頭症
2.特徴的な鳥様顔貌
3.低身長
4.免疫不全による易感染性
5.放射線感受性の亢進
リンパ系悪性腫瘍、固形腫瘍の合併が高率である。
B.重要な検査所見
1.T細胞数の低下
2.B細胞数の低下、IgGサブクラスとIgA、IgEの低下、IgMの上昇
3.放射線高感受性
リンパ球と線維芽細胞の染色体不安定性
確定診断には、NBS1(Nibrin)遺伝子変異を同定する。常染色体劣性遺伝形式をとる。
<IV.ブルーム(Bloom)症候群>
診断方法:
A.主要臨床症状
1.小柄な体型
2.特徴的な鳥様顔貌
3.日光過敏性紅斑
4.造血不全
5.放射線感受性の亢進
造血器腫瘍(白血病、リンパ腫)の合併が高率である。
6.糖尿病の合併
7.不妊
B.重要な検査所見
1.上記の症状が認められた場合は、姉妹染色体分体の交換(sister chromatid exchange)の頻度を解析する。ブルーム症候群では、sister chromatid exchangeの頻度の上昇が認められる。
2.T細胞数は正常。
3.B細胞数は正常。免疫グロブリン値の低下。
確定診断には、DNAヘリカーゼをコードするBLM遺伝子変異を同定する。常染色体劣性遺伝形式をとる。
<V.ICF症候群>
診断方法:
A.主要臨床症状
1.特徴的顔貌
眉間解離、低位耳介、巨舌
2.易感染性
3.栄養吸収不全
B.重要な検査所見
1.T細胞数は減少あるいは正常
2.B細胞数は減少あるいは正常
3.低ガンマグロブリン血症を呈する。
確定診断として、DNAメチル化に重要なDNAメチルトランスフェラーゼ-3bをコードするDNMT3B遺伝子変異を同定する。常染色体劣性遺伝形式をとる。
<VI.PMS2異常症>
診断方法:
A.主要臨床症状
1.易感染性による反復性感染症
2.カフェオレ班
3.悪性腫瘍の高頻度合併
造血器腫瘍、大腸癌、脳腫瘍、その他
B.重要な検査所見
1.T細胞数は正常
2.B細胞数の減少
3.IgGとIgAの低下、IgMの上昇
免疫グロブリンクラススイッチ異常による。
確定診断には、DNAミスマッチ修復に重要なPMS2遺伝子異常を同定する。常染色体劣性遺伝形式をとる。
類縁疾患概念としてリンチ症候群があり、DNAミスマッチ修復遺伝子群(MLH1、MSH2、MSH6、PMS2)の生殖細胞系列の変異による遺伝性疾患である。
<VII.RIDDLE症候群>
診断方法:
A.主要臨床症状
1.放射線高感受性
2.免疫不全による易感染性
3.特徴的顔貌
4.学習障害
B.重要な検査所見
DNA二重鎖損傷に対する修復機構として、ATMや制御因子の凝集体形成が必要であるが、これらのDNA損傷部位への凝集体リクルートが欠損している。
確定診断として、RING型E3ユビキチンリガーゼをコードするRNF168遺伝子異常を同定する。
常染色体劣性遺伝形式をとる。
<VIII.シムケ(Schimke)症候群>
診断方法:
A.主要臨床症状
1.骨格系異形成による低身長、子宮内発育不全
2.不均衡体型
3.顔貌異常
4.腎障害
5.細胞性免疫不全による易感染性
6.造血不全
B.重要な検査所見
1.T細胞数の減少
2.B細胞数及び免疫グロブリン値は正常
3.確定診断として、染色体リモデリングに重要なSMARCAL1遺伝子変異を同定する。常染色体劣性遺伝形式をとる。
<IX. ネザートン(Netherton)症候群>
診断方法:
A.主要臨床症状
1.先天性魚鱗癬
乳児期より発症する。
2.毛髪異常
頭髪はまばらで短く、もろい。体毛も異常である。
3.アトピー体質
蕁麻疹、血管性浮腫、アトピー性皮膚炎、喘息
4.発育不良
5.易感染性
6.一部で精神発達遅滞
B.重要な検査所見
1.T細胞数は正常
2.B細胞数は減少、血清IgEの上昇
3.NK細胞機能低下
4.確定診断として、上皮系細胞に発現するセリンプロテアーゼインヒビターをコードするLEKT1遺伝子変異を同定する。常染色体劣性遺伝形式をとる。
<X.胸腺低形成(DiGeorge症候群、22q11.2欠失症候群)>
診断方法:
A.主要臨床症状
1.副甲状腺低形成による低カルシウム血症による症状
2.胸腺低形成による易感染性
3.心流出路奇形
ファロー四徴症、円錐動脈管心奇形、大動脈弓離断、右大動脈弓、右鎖骨下動脈起始異常等の心奇形など
4.特異的顔貌
口蓋裂、低位耳介、小耳介、瞼裂短縮を伴う眼角隔離症、短い人中、小さな口、小顎症など
5.精神発達遅滞、言語発達遅滞
B.重要な検査所見
1.低カルシウム血症、副甲状腺機能低下
2.T細胞数は減少及び機能低下
3.B細胞数は正常、免疫グロブリン値は正常か減少
4.画像検査や心カテーテルによる心奇形の同定
5.確定診断として、微細染色体欠失症候群として染色体22q11.2 の微細欠失をfluorescence in situ hybridization(FISH)やarray comparative genomic hybridization(aCGH)にて同定する。特に、TBX1遺伝子のハプロ不全が、身体的奇形の出現に大きな役割を演ずるとされる。常染色体優性遺伝形式かde novo遺伝形式をとる。
<XI.高IgE症候群>
診断方法:
A.主要臨床症状:
1.黄色ブドウ球菌を中心とする細胞外寄生細菌による皮膚膿瘍と肺炎
2.新生児期から発症するアトピー性皮膚炎
3.血清IgEの高値
を3主徴とする。
1型と2型があり、1型の多くの症例で特有の顔貌、脊椎の側弯、病的骨折、骨粗鬆症、関節の過伸展、乳歯の脱落遅延などの骨・軟部組織・歯牙の異常を合併する。2型は、さらに、細胞内寄生細菌とウイルス(単純ヘルペスウイルス、伝染性軟属腫)に対する易感染性、中枢神経合併症が見られる。
B.重要な検査所見
1.T細胞数は正常だが、Th17細胞は減少する。
2.B細胞数は正常だが、特異的抗体産生は低下する。
3.血清IgEの高値
4.画像検査にて慢性呼吸器感染像と肺嚢胞
5.骨密度の低下
6.確定診断として、1型高IgE症候群は片アリルのSTAT3遺伝子異常を同定するが、主に散発性でありまれに常染色体優性遺伝形式をとることがある。2型高IgE症候群はTYK2遺伝子異常を同定するが、主に常染色体劣性遺伝形式を呈する。
<XII.肝中心静脈閉鎖症を伴う免疫不全症>
診断方法:
A.主要臨床症状
1.肝中心静脈閉鎖
2.肝脾腫
3.反復する呼吸器感染
4.血小板減少
B.重要な検査所見
1.記憶T細胞の低下
2.記憶B細胞の低下
3.画像検査にて肝中心静脈閉鎖の所見
4.確定診断として、細胞核に発現するSP110遺伝子変異を同定する。常染色体劣性遺伝形式をとる。
<XIII.先天性角化不全症>
診断方法:
テロメア長の維持機能の障害を背景とし、主に皮膚、爪、口腔粘膜に特徴的な所見を有する遺伝子骨髄不全症候群である。古典的な先天性角化不全症の他に最重症型であるHoyeraal-Hreidarsson症候群、Revesz症候群の他、不全型である再生不良性貧血や家族性肺線維症などが存在する。
A.主要臨床症状
狭義な意味での先天性角化不全症は、骨髄不全及び1つ以上の大症状と2つ以上の小症状を満たす場合に診断する。
1.骨髄不全症
一系統以上の血球減少と骨髄低形成を認める。
2.大症状(皮膚、粘膜所見)
1)網状色素沈着
2)爪の萎縮
3)口腔粘膜白斑症
3.小症状(その他の身体所見)
1)頭髪の消失、白髪
2)歯牙の異常
3)肺病変
4)低身長、発達遅延
5)肝障害
6)食道狭窄
7)悪性腫瘍
8)小頭症
9)小脳失調
10)骨粗鬆症
B.重要な検査所見
1.T細胞数の減少
2.B細胞数の減少
3.NK細胞数の減少と機能低下
4.汎血球減少
5.確定診断として、染色体テロメア長の制御に重要な遺伝子群の変異を同定する。X連鎖性遺伝形式をとるDKC1(dyskerin)、常染色体性形式をとるTERC、TERT、NHP2、NOP10、TINF2遺伝子などの変異を同定する。
③液性免疫不全を主とする疾患
<I.X連鎖無ガンマグロブリン血症>
診断方法
1.男児に発症
2.生後4~8か月頃から感染症にかかりやすくなる
3.血清免疫グロブリン値著減(IgG<200mg/dL、 IgA及びIgMは感度以下)
4.末梢血B細胞欠損(<2%)
5.扁桃、リンパ節は痕跡程度
6.細胞性免疫能は正常
7.家族歴(兄弟、母方従兄弟またはおじ)
8.BTK遺伝子変異またはBTK蛋白欠損
・女児においても発症し、臨床像及び検査所見から区別しがたい常染色体劣性無ガンマグロブリンが存在する。その原因遺伝子としてμ重鎖、Igα、Igβ、λ5、BLNKがある。
<II.分類不能型免疫不全症>
診断方法
1.血清IgGの著明な低下を示し、IgA及びIgMの低下を伴う
2.予防接種に対する反応の低下または欠損
3.その他の免疫不全症がないこと
・TACI、 ICOS、 BAFF-R、 CD19、 CD81、 CD20、 CD21変異例が報告されている
<III.高IgM症候群>
診断方法
1.血清IgG、 IgA、 IgEの欠損を伴う
2.血清IgMは正常又は高値
・CD40リガンド(CD154)変異によるX連鎖高IgM症候群が最も多いが、常染色体劣性高IgM症候群としてCD40、 AICDA又はAID、 UNG変異によるものもある。
<IV.IgGサブクラス欠損症>
診断方法
1.反復性の重症感染症を呈する
2.1つ又はそれ以上のIgGサブクラス欠損
3.トータルのIgGは正常か正常に近い濃度である
<V.選択的IgA欠損症>
診断方法
1.血清IgAのみが低下(血清IgG及びIgMは正常)
2.4歳以上(4歳以下では血清IgAが正常化するまで経過観察が必要である)
3.低ガンマグロブリン血症を呈する他の疾患が除外されている
<VI.特異抗体産生不全症>
診断方法
1.多糖体ワクチンに対する反応が低下
2.IgG、IgGサブクラス、IgA、IgM、IgEは正常
3.その他の原発性又は二次性原発性免疫不全症が除外されている
<VII.乳児一過性低ガンマグロブリン血症>
診断方法
1.血清IgGが年齢相応の正常値の-2SD未満である
2.その他の血清免疫グロブリンの値は問わない
3.生後6か月以降
4.その他の原発性免疫不全症が除外されている
<VIII.その他の液性免疫不全を主とする疾患>
・モノソミー7、トリソミー8、先天性角化不全症による低ガンマグロブリン血症を伴う骨髄異形成がある。
・1つ又はそれ以上のIgG及びIgAサブクラスの低値を伴う、免疫グロブリン重鎖の変異又は欠失がある。
④免疫調節障害
<I.チェディアック・東(Chédiak-Higashi)症候群>
【診断方法】
A.症状
1.皮膚、毛髪、眼における部分的白子症
2.一般化膿菌に対する易感染性
3.知能障害、痙攣、小脳失調、末梢神経障害等の神経系の異常(ただし、幼少期には目立たず、進行性)
4.出血傾向
5.血球貪食症候群の合併
B.検査所見
1.白血球内の巨大顆粒(ミエロペルオキシダーゼや酸フォスファターゼが陽性)
2.NK細胞活性の低下
3.細胞傷害性T細胞の機能障害
4.LYST遺伝子変異
・病的なLYST遺伝子変異が認められれば、確定診断される。
・部分的白子症を伴う先天性免疫不全症で、白血球内の巨大顆粒を認める場合、本症の可能性が高い。
・類縁疾患にGricelli症候群、ヘルマンスキー・パドラック(Hermanski-Pudlak)症候群が知られている。
<II.X連鎖リンパ増殖症候群>
【診断方法】
A.症状
1.EBウイルスによる致死的伝染性単核症
2.血球貪食症候群
3.低ガンマグロブリン血症
4.SAP欠損症では、悪性リンパ腫、再生不良性貧血、血管炎
5.XIAP欠損症では、脾腫、出血性腸炎
B.検査所見
1.リンパ球におけるSAP又はXIAP蛋白発現の低下
2.SH2D1A又はXIAP/BIRC4遺伝子の変異
3.インバリアントNKT細胞の低下
・XLPには、タイプ1のSAP欠損症とタイプ2のXIAP欠損症が知られている。
・原則として男児に発症する。
・SH2D1A又はXIAP/BIRC4遺伝子に病的な変異が認められれば、確定診断される。
・男児で重症のEBウイルス感染症を発症、又は血球貪食症候群を繰り返す場合には、本症を疑う。
<III.自己免疫性リンパ増殖症候群(ALPS)>
【診断基準】
A.必須項目
1.6か月を超えて慢性に経過する非腫瘍性、非感染性のリンパ節腫脹若しくは脾腫、又はその両方
2.CD3+ TCRαβ+ CD4– CD8– T細胞(ダブルネガティブT細胞)の増加(末梢血リンパ球数が正常又は増加している場合で、全リンパ球中の1.5%以上又はCD3+ T細胞の2.5%以上)
B.付帯項目
1.一次項目
1) リンパ球のアポトーシスの障害(2回の独立した検索が必要)
2) FAS、FASLG、CASP10のいずれかの遺伝子における体細胞又は生殖細胞系列での変異
2.二次項目
1) 血漿sFASL(>200 pg/mL)、血漿IL-10(>20 pg/mL)、血清又は血漿ビタミンB12(>1500 ng/L)、血漿IL-18(>500 pg/mL)のいずれかの増加
2) 典型的な免疫組織学的所見(経験豊富な血液病理学者による)
3) 自己免疫性血球減少(溶血性貧血、血小板減少又は好中球減少)かつ多クローン性IgGの増加
4) 自己免疫の有無にかかわらず非腫瘍性/非感染性のリンパ球増殖症の家族歴
・必須項目2つと付帯項目の一次項目1つを満たせば、確定診断される。
・必須項目2つと付帯項目の二次項目1つを満たせば、本症の可能性が高い。
・類縁疾患にカスペース8欠損症、RAS関連自己免疫性リンパ増殖症候群様疾患(RALD)、FADD欠損症が知られている。
<IからIIIに掲げるもののほか、免疫調節障害>
【診断方法】
その他の免疫調節障害として、家族性血球貪食症候群(FHL)、カンジダ感染と外胚葉形成異常を伴う自己免疫性多腺性内分泌不全症(APECED)、IPEX症候群、CD25欠損症、ITCH欠損症などが知られている。
家族性血球貪食症候群(FHL)では、症状や一般検査から他の原因による血球貪食症候群とFHLを鑑別することは困難である。FHLの病型には、FHL1(原因遺伝子不明)、FHL2(パーフォリン欠損症)、FHL3(Munc13-4欠損症)、FHL4(Syntaxin11欠損症)、FHL5(Munc18-2欠損症)が知られている。FHL2~FHL5では、それぞれの原因遺伝子の変異が認められれば、確定診断される。また、それぞれの蛋白発現解析によるスクリーニングが可能である。NK細胞活性や細胞傷害性T細胞の機能は一般に低下する。
APECEDは内分泌症候群、IPEX症候群は慢性消化器症候群の項を参照
⑤原発性食細胞機能不全症及び欠損症
<I.重症先天性好中球減少症>
1.生後早期からの反復する重症細菌感染症
2.慢性好中球減少(末梢血好中球絶対数が200/mL未満)
3.骨髄像で骨髄顆粒球系細胞の正形成~低形成と前骨髄球を認める
4.既知の遺伝子として、ELANE、HAX1、GFI1、CSF3R、WAS、G6PC3が挙げられる
・好中球エラスターゼをコードするELANE遺伝子の変異が約60%
・その他に、HAX1遺伝子やGFI1遺伝子、G−CSF受容体であるCSF3R遺伝子の変異、Wiskott-Aldrich Syndrome protein(WAS)の恒常活性型変異、先天性心疾患、静脈拡張、泌尿生殖器異常を伴うG6PC3遺伝子異常がある。
<II.周期性好中球減少症>
1.約21日周期での好中球減少
2.周期に一致した発熱、口内炎、全身倦怠感
3.3~5日で自然回復する。
4.好中球減少(末梢血好中球絶対数が500/µL未満)
5.ほぼ全例で好中球エラスターゼ遺伝子(ELANE)変異が認められる。
・末梢血での血液検査に先行し骨髄像の変化(低形成~過形成)が見られるが、周期によって違うため骨髄像からの診断は難しい。
<III.I及びIIに掲げるもののほか、慢性の経過をたどる好中球減少症>
その他に慢性的な経過をたどる好中球減少症として様々な責任遺伝子が明らかになっており、今後も増えることが予想される。代表的なものとして、ヘルマンスキー・パドラック(Hermanski-Pudlak)症候群2型(AP3B1)、Griscelli症候群2型(RAB27A)、p14欠損症(P14/MAPBPIP)、WHIM症候群(CXCR4)や糖原病Ib型(G6PT1)などが挙げられる。責任遺伝子を括弧内に示す。
<IV.白血球接着不全症>
LADタイプI:b2インテグリンの欠損による接着障害
1.生後早期からの細菌感染症
2.非化膿性の皮膚感染症、臍帯脱落遅延、歯肉炎、歯周囲炎
3.白血球異常高値
4.粘着能、遊走能、貪食能の低下
5.フローサイトメトリーによるCD18、CD11の欠損にて診断される。
6.責任遺伝子はINTGB2である。
LADタイプIIはセレクチンリガンドのフコシル化炭水化物欠損による接着障害であり、LADタイプIの症状に加えて精神発達遅滞が認められる。LADタイプIIIはLADタイプIの症状に加えて出血症状があり、b2インテグリンと相互作用するKindlin-3の欠損により生ずる。責任遺伝子はそれぞれFUCT1(タイプII)とKINDLIN3(タイプIII)であるが、頻度は極めて低い。
<V.シュワッハマン・ダイアモンド(Shwachman-Diamond)症候群>
1.常染色体劣性遺伝
2.好中球減少症による易感染性、貧血、血小板減少
3.膵眼分泌異常
4.骨格異常(低身長など)を伴うことが多い
5.骨髄異形成症候群、急性骨髄性白血病を発症することが多い
6.90%でSBDS遺伝子に変異が認められる
上記臨床症状のもとSBDS遺伝子解析により確定診断にいたる。
<VI.慢性肉芽腫症>
活性酸素産生好中球が正常コントロールの5%未満で、下記のうち1つを満たす。
1.難治性深部感染症(カタラーゼ陽性菌、真菌等)の罹患歴
2.皮膚、リンパ節、肺、消化管、尿路系などのびまん性肉芽腫形成
3.食細胞殺菌能低下がNBT還元能またはDHR-123法にて認められた場合
上記臨床症状及び検査結果のもと以下の遺伝子解析により確定診断にいたる。
gp91phox、p22phox、p47phox、p67phox、p40phoxの異常により活性酸素産生能が低下することもある。
<VII.ミエロペルオキシダーゼ欠損症>
1.常染色体劣性遺伝
2.好中球の細胞内殺菌能低下
3.カンジダ症罹患(5%未満)
4.好中球のMPO染色によるMPO欠損、減少
5.偶然発見され、無症状の症例も多い
上記臨床症状のもとMPO遺伝子解析により確定診断にいたる。
<VIII.メンデル遺伝型マイコバクテリア易感染症>
1.BCG、非結核性抗酸菌に対する易感染性
2.サルモネラ等の細胞内寄生菌感染症による重篤化
3.多発性骨髄炎
4.他の感染症に対しては易感染性を示さない
上記臨床症状のもと以下の遺伝子解析により確定診断にいたる。
IL12B、IL12RB1、IFNGR1、IFNGR2、STAT1、IKBKG、CYBB、TYK2、IRF8、ISG15
<IVからVIIIに掲げるもののほか、白血球機能異常>
白血球機能異常を示す上記以外の疾患
⑥自然免疫異常
I.免疫不全を伴う無汗性外胚葉形成異常症
II.IRAK4欠損症
III.MyD88欠損症
IV.慢性皮膚粘膜カンジダ症
V. IからIVに掲げるもののほか、自然免疫異常
診断方法
自然免疫において重要な役割を果たす分子の先天的な欠損あるいは機能異常があり、それによる自然免疫機構の障害によって易感染性を呈する疾患であり、多くの場合、その分子の欠損あるいは機能異常に直接的に関連する遺伝子異常が認められる。
診断は、各疾患の特徴的な臨床像に加えて、以下のいずれかがある場合を原則とする。
1.該当する分子の欠損が証明できる場合
2.該当する遺伝子異常が、該当する分子の欠損や機能異常に結び付くことが直接的に証明できる場合
3.該当する分子や責任遺伝子の異常がない、あるいは原因が解明されていないが、該当する疾患の病態の根本的な基盤となる現象を、免疫学的あるいは分子生物学的手法を用いて証明できる場合
4.易感染性が、該当する疾患以外では医学的に説明できない場合
この疾患は、以下のように細分類される。細分類ごとに、上記の方法によって診断する。
現在判明している責任遺伝子を各々括弧内に示す。
免疫不全を伴う無汗性外胚葉形成異常症(IKBKG、IKBA)
無汗性外胚葉形成異常と種々の病原体に対する易感染性を特徴とする。無汗症や外胚葉形成不全の症状、易感染性の程度は様々である。
IRAK4欠損症(IRAK4)
肺炎球菌、ブドウ球菌、連鎖球菌、緑膿菌などによる侵襲性細菌感染症を特徴とする。特に肺炎球菌による化膿性髄膜炎は死亡率が高い。
MyD88欠損症(MYD88)
IRAK4欠損症と臨床像は類似している。
慢性皮膚粘膜カンジダ症(IL17RA、IL17F、STAT1、ACT1)
皮膚や粘膜、爪の慢性的なカンジダ症を呈する疾患である。抗真菌剤は一時的に有効であるが、長期的に完全に病変を治癒させることは困難である。通常深部臓器の真菌症は伴わない。
他の自然免疫異常
これには、WHIM(warts, hypogammaglobulinemia, infections, myelokathexis)症候群、疣贅様表皮発育異常症(Epidermodysplasia verruciformis)、単純ヘルペス脳炎、CARD9欠損症があり、それぞれ、CXCR4、EVER1/ EVER2、TLR3/ UNC93B1/ TRAF3/ TRIF/ TBK1、CARD9、APOL-1が責任遺伝子である。
⑦先天性補体欠損症
I.先天性補体欠損症
II.遺伝性血管性浮腫(C1インヒビター欠損症)
III.Ⅰ及びⅡに掲げるもののほか、先天性補体欠損症
診断方法
補体は30種類以上の様々な機能をもつ分子群であり、先天的な欠損による臨床症状は様々である。大きく分類すると、
1.前期反応経路の異常
2.後期反応経路の異常
3.制御因子、及びその受容体の異常
に分けられる。1では、欠損する補体成分に関連した易感染性だけでなく、全身性エリテマトーデス類似の自己免疫疾患が起こりやすい。2では、ナイセリア属に対する易感染性が見られるが、全身性エリテマトーデスなどの自己免疫疾患の頻度は少ない。C9欠損症は日本人で頻度が高いが、髄膜炎菌による化膿性髄膜炎の頻度が正常人よりも高いとされる。3には、C1インヒビター欠損による遺伝性血管浮腫、及びFactor IやFactor H、MCPなどの第2経路の異常によるものがあり、後者は非典型溶血性尿毒症症候群(aHUS)の原因となる。ここでは、aHUSや、補体系による溶血を呈する発作性夜間欠色素尿症については、他のカテゴリーに属するものとする。補体欠損症には胎生期の細胞の遊走能異常を起こすものもある。
補体欠損症は以下のように細分類される。
・先天性補体欠損症
先天性補体欠損症は、以下のように更に細分類される。現在判明している責任遺伝子を各々括弧内に示す。診断は、補体成分の欠損を証明するか、対応する責任遺伝子にそれに直接関連した異常を認めることで診断する。なお、感染症や自己免疫疾患等に付随して起こる補体の消費等による二次的な補体成分の低下は、この疾患に含めてはならない。
C1q欠損症(C1QA、C1QB、C1QC)、C1r欠損症(C1R)、C1s欠損症(C1S)、C4欠損症(C4A、C4B)、C2欠損症(C2)、C3欠損症(C3)、C5欠損症(C5A、C5B)、C6欠損症(C6)、C7欠損症(C7)、C8欠損症(C8A、C8B)、C9欠損症(C9)、Factor D欠損症(CFD)、Properdin欠損症(PFC)、Factor I欠損症(CFI)、Factor H欠損症(CFH)、MASP1欠損症(MASP1)、3MC症候群(CLK1)、MASP2欠損症(MASP2)、Ficolin 3関連免疫不全症(FCN3)
・遺伝性血管性浮腫
これには以下の3つの病型が含まれる。
1型:C1インヒビターの活性、蛋白量ともに低下している。
2型:C1インヒビターの活性は低下しているが、蛋白量は正常又は上昇している。
3型:遺伝性であるが、C1インヒビターの活性、蛋白量ともに正常である。
診断は、遺伝性血管性浮腫の臨床像をもとに、C1インヒビター活性を測定し、正常値の70%以下であれば、家族歴を問わず、遺伝性血管性浮腫と診断する。なお、発作時のC4値の低値は診断の参考となる。3型は極めてまれであるが、典型的な臨床像を呈し、家族性に認められれば、C1インヒビター活性が低値でなくても、遺伝性血管性浮腫と診断して良い(これまで国内からは報告されていない。)。
・ほかの先天性補体欠損症
特徴的な臨床像を呈し、補体成分の欠損とそれに直接関連した責任遺伝子の異常が確認できれば診断する。
<重症度分類>
原発性免疫不全症候群全体について、中等症以上を対象とする。
重症
治療で、補充療法(阻害薬等の代替治療薬の投与を含む。)、G-CSF療法、除鉄剤の投与、抗凝固療法、ステロイド薬の投与、免疫抑制薬の投与、抗腫瘍薬の投与、再発予防法、感染症予防療法、造血幹細胞移植、腹膜透析、血液透析のうち、1つ以上を継続的に実施する(断続的な場合も含めて概ね6か月以上)場合。
中等症
上記治療が継続的には必要でない場合。
軽症
上記治療が不要な場合。
※診断基準及び重症度分類の適応における留意事項
1.病名診断に用いる臨床症状、検査所見等に関して、診断基準上に特段の規定がない場合には、いずれの時期のものを用いても差し支えない(ただし、当該疾病の経過を示す臨床症状等であって、確認可能なものに限る。)。
2.治療開始後における重症度分類については、適切な医学的管理の下で治療が行われている状態であって、直近6か月間で最も悪い状態を医師が判断することとする。
3.なお、症状の程度が上記の重症度分類等で一定以上に該当しない者であるが、高額な医療を継続することが必要なものについては、医療費助成の対象とする。
- 一般社団法人 日本免疫不全・自己炎症学会HP:https://www.jsiad.org/
- NPO法人PIDつばさの会:https://npo-pidtsubasa.org/
| 研究班名 | 原発性免疫不全症候群の全国診療体制確立、移行医療体制構築、診療ガイドライン確立に関する研究班 研究班名簿 |
|---|---|
| 情報更新日 | 令和4年12月(名簿更新:令和7年6月) |
治験を実施している指定難病を調べることができます。キーワード検索欄に指定難病名を入力し、検索してください。
治験・臨床研究情報検索サイト一覧