三尖弁閉鎖症(指定難病212)
1. 「三尖弁閉鎖症」とは
正常な心臓では図1左のように、全身の静脈血は上・下大静脈から右房へ戻り、右室、肺動脈、肺へと流れ、酸素が豊富な血液となって肺静脈から左房へ戻り、左室、大動脈の順に流れていきます。肺で酸素を取り込むため、肺静脈はもっとも多くの酸素を含んでいます。右房と右室の間にある弁を三尖弁と呼びます。
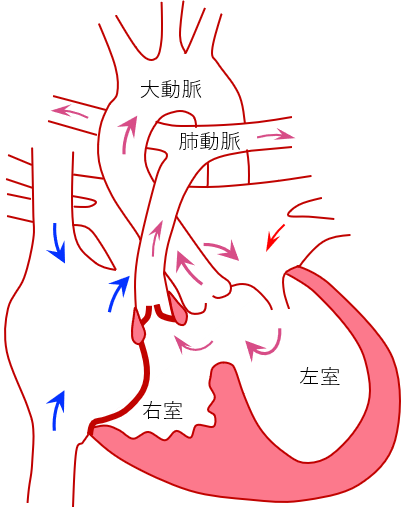
図1:三尖弁閉鎖症(Ib型)
三尖弁閉鎖症は、生まれつき三尖弁が閉鎖している病気です。そのため、右房へ戻ってきた静脈血は右室に流れ込むことができず、すべて心房間の孔(心房中隔欠損または卵円孔)を通って左房へ流れ込み、左房の血液と混合し、僧帽弁を通って左室へ流れ込みます。正常では左房の血液は多くの酸素を含んでいますが、そこへ酸素の少ない静脈血が流れ込むため、酸素の含有量が低下します。この血液が左心室、大動脈を通って体に送られるため、 チアノーゼ が見られることになります(図1右)。この病気はチアノーゼを主症状とする先天性心臓病のなかで3番目に多い病気です。右室は小さいことがほとんどで、心臓手術を行っても通常の右室として使用することはできません。従って、使用できる心室が左心室のみであり、単心室症と同じように最終的にフォンタン型の手術を目指すことになります。
三尖弁閉鎖症は大きく分けると、肺へ流れる血液量が少なく、チアノーゼ(低酸素血症)が主な症状となるタイプ(肺血流減少型)と、肺へ流れる血液量が多いために肺や心臓に負担がかかり、呼吸器症状、肝臓の腫れ、むくみ、体重増加不良などの心不全が目立つタイプ(肺血流増加型)があり、治療、外科手術の経過も異なりますが、最終的にフォンタン型手術を目指すことは同じです。
2. この病気の患者さんはどのくらいいるのですか。
三尖弁閉鎖症の発症率は先天性心疾患のおよそ1%(出生10,000人に対して1人)の割合と言われています。最近の日本の出生数は年間約100万人ですので、毎年、全国で100人程度の三尖弁閉鎖症の患者さんが生まれていることになります。成人になった三尖弁閉鎖症がどのくらいいるかは、まだ明らかではありません。外科治療の進歩により毎年増えていることが予想されます。
3. この病気はどのような人に多いのですか。
ほとんどの場合は様々な原因が関与して発症するため、特にどのような人に多いという傾向は明らかではありません。しかし、ときに同一家系内に発生する例、染色体異常に伴って発生する例がみられます。三尖弁閉鎖症の約10~15%では心臓以外の先天性異常を伴っています。
4. この病気の原因はわかっているのですか。
胎児期(胎生30日ころ)に右心房と右心室、左心房と左心室の繋がりができあがっていく過程がうまく行かず、三尖弁口が閉鎖してしまいますが、その原因は不明です。
5. この病気は遺伝するのですか。
ほとんどの場合、遺伝はしませんが、ときに 家族性 にみられる場合があります。なんらかの遺伝子変異が関係している可能性はありますが、まだ明確ではありません。
6. この病気ではどのような症状がおきますか。
三尖弁閉鎖症の型や合併する他の構造異常などによって異なりますが、肺へ流れる血液量が少ない肺血流減少型ではチアノーゼ(低酸素血症)が主な症状となります。出生後、動脈管が自然に閉じはじめると肺血流量はその分減少し、チアノーゼは次第に強くなります。また、右心室から肺動脈への出口(右室流出路と呼ぶ)の 狭窄 が進行して、ファロー四徴症と同様の低酸素発作を生じることがあります。低酸素発作の症状は、チアノーゼの増強、多呼吸、意識障害などです。一方、肺血流増加型ではチアノーゼは目立ちませんが、肺へ流れる血液量が多いために肺や心臓が負担を受け、呼吸障害、肝臓の腫大、浮腫、体重増加不良などの うっ血 性心不全の症状が目立ちます。
7. この病気にはどのような治療法がありますか?
チアノーゼが進行するときは肺へ流れる血液量の不足が主な原因であるため、酸素を吸入しても良くなりません。プロスタグランジンという薬を持続静注して動脈管を閉じないようにして、肺へ流れる血液量を確保する治療を行います。動脈管は大動脈と肺動脈を橋渡ししている血管で、出生後に自然に閉鎖する性質があります。これを開存させることによって肺血流を維持することができるのです。その後、2~4週間のうちに動脈管を開存させておく代わりに シャント手術 を行います。
逆に肺血流が多すぎて心不全症状があるときは、肺動脈絞扼術(肺動脈の回りにテープをかけて絞り込む手術)を行って、肺へ流れる血液量を減らします。また、心房間の交通が不十分で、静脈がうっ血するときは、バルーンカテーテルを使って卵円孔を大きくする カテーテル治療 (心房中隔裂開術:BASと呼ぶ)が必要となることもあります。
三尖弁閉鎖症では右心室は小さくて使用できないので、単心室と同様の考え方でチアノーゼをなくすことを目標として、最終的にフォンタン型の手術を目指します。しかし、ほとんどの症例ではフォンタン型手術を行う前にいくつかの段階的手術が必要となります。両方向性グレン手術(上大静脈を肺動脈につないで、まず上半身の静脈のみ肺へ流す手術)もその一つです。また、すべての患者さんにフォンタン型手術ができるわけではなく、肺動脈が細すぎるなど様々な理由で、フォンタン型手術に到達できないことがあります。
8. この病気はどういう経過をたどるのですか。
タイプによっても経過は違いますが、手術を行わない場合の生存率は1歳で約60%、10~15歳に達するのは半数以下と言われています。フォンタン型手術が成立すれば生存率は改善しますが、そこに到達するまでに段階的な手術を必要とすることが多く、フォンタン型手術は1~3歳前後で行われることが多いようです。しかし、フォンタン型手術に到達して経過の良好な患者さんでも、術後10~20年以上すると、フォンタン循環という特殊な血液の流れに伴う様々な合併症(フォンタン術後症候群)が出現してきます。フォンタン型手術は決して病気を治す手術ではなく、あくまで低酸素状態を回避するために行うものであり、やはり、一つの心室で一生涯、全身の循環を維持することにはどこか無理があるようです。特に 予後 に影響する主な合併症は、心不全、不整脈、うっ血肝(フォンタン術後肝疾患)、 血栓症 ですが、それ以外にも、低酸素血症(チアノーゼ)の再発、腎障害、 蛋白漏出性胃腸症 、特殊な気管支炎など様々です。
9. この病気は日常生活でどのような注意が必要ですか。
フォンタン型手術を行った患者さんでは上記の合併症に注意して生活することが重要です。心不全の症状としては、顔や手足のむくみ、お腹の張り、息切れや動悸、持続する咳や痰、夜間の息苦しさなどがあります。不整脈の症状としては、動悸、胸痛、めまいなどがありますが、特に失神(意識消失)のあったときは要注意です。これらの症状があったときには早期に病院を受診するようにします。また、血栓症の誘因となる脱水症にも気を付け、血栓予防の薬についても主治医とよく相談し、定期的に血液検査を受け、薬の効き具合を調べるようにします。運動については、激しい運動は困難なことが多いですが、自分に合った適度な身体的活動、運動を無理しない程度に日常に取り入れて体力を付けることも重要です。
女性では妊娠と出産のことも問題となります。フォンタン型手術後の女性では妊娠中に心臓に負担がかかり、危険を伴うことが少なくありません。血栓予防のためにワルファリンを内服している場合は妊娠は禁止となります。妊娠可能な年齢になったら、早めから主治医の先生、成人先天性心疾患専門医、産科の先生とよく相談して十分な知識を持っておくことが必要です。
10. 次の病名はこの病気の別名又はこの病気に含まれる、あるいは深く関連する病名です。 ただし、これらの病気(病名)であっても医療費助成の対象とならないこともありますので、主治医に相談してください。
該当する病名はありません。
11. この病気に関する資料・関連リンク
① 小児・成育循環器学(改訂第2版). 日本小児循環器学会編集. 診断と治療社, 2024.
② 日本成人先天性心疾患学会ホームページ総合・連携修練施設一覧
https://www.jsachd.org/specialist/list-facility/
③ 成人先天性心疾患診療ガイドライン(2025年改訂版)日本循環器学会.
https://www.j-circ.or.jp/cms/wp-content/uploads/2025/03/JCS2025_Yamagishi.pdf
④ 先天性心疾患並びに小児期心疾患の診断検査と薬物療法ガイドライン(2018年改訂版)日本循環器学会.
https://www.j-circ.or.jp/cms/wp-content/uploads/2020/02/JCS2018_Yasukochi.pdf
⑤ 心疾患患者の妊娠・出産の適応、管理に関するガイドライン(2018年改訂版)日本循環器学会.
https://www.j-circ.or.jp/cms/wp-content/uploads/2018/06/JCS2018_akagi_ikeda.pdf
⑥ 先天性心疾患術後遠隔期の管理・侵襲的治療に関するガイドライン(2022年改訂版)日本循環器学会.
https://www.j-circ.or.jp/cms/wp-content/uploads/2022/03/JCS2022_Ohuchi_Kawada.pdf
治験を実施している指定難病を調べることができます。キーワード検索欄に指定難病名を入力し、検索してください。
治験・臨床研究情報検索サイト一覧
| 研究班名 | 先天性心疾患を主体とする小児期発症の心血管難治性疾患の救命率の向上、円滑な移行医療、成人期以降の予後改善を目指した総合的研究班 研究班名簿 |
|---|---|
| 情報更新日 | 令和7年12月(名簿更新:令和7年6月) |