混合性結合組織病(指定難病52)
○ 概要
1.概要
混合性結合組織病(mixed connective tissue disease:MCTD)は、全身性エリテマトーデス、全身性強皮症、及び、多発性筋炎/皮膚筋炎などに認められる臨床症候や検査所見が同時にあるいは経過とともに混在し、血清中に抗U1-RNP(ribonucleoprotein)抗体が検出される疾患である。
2.原因
MCTDは全身性自己免疫疾患の一つである。明確な原因は特定されていないが、疾患に特徴的な免疫異常は、高力価に認められる抗U1-RNP抗体である。同抗体の産生はHLAに拘束されることが知られ、遺伝的な素因が関係するとともに、一部のウイルス感染など環境要因の関与も推測されている。
3.症状
レイノー現象が大半の症例で見られる。MCTDではレイノー現象と「手指または手背の腫脹」が、いつまでも長く持続することが特徴的である。このためこれらの症状は、MCTDに特徴的な共通症状として重視され、多くの例での初発症状となっている。
MCTDに特徴的な所見として、肺動脈性肺高血圧症、無菌性髄膜炎、三叉神経障害が挙げられる。肺動脈性肺高血圧症は、10-50%に認められ、重篤な病態であり、早期に発見して適切な治療が必要となる。MCTDと診断されたら、肺動脈性肺高血圧症の有無について心臓超音波検査や右心カテーテル検査などを行う。三叉神経障害は三叉神経Ⅱ枝又はⅢ枝のしびれ感を主体とした症状で、MCTDの約10%にみられる。無菌性髄膜炎も本症では約10%にみられ、原疾患の活動性に伴うものと非ステロイド抗炎症薬(NSAIDs)などの薬剤に起因するものがある。ただし、特徴的な臓器病変については十分な鑑別診断を要する。たとえば、無菌性髄膜炎をきたす疾患として高頻度な感染性髄膜炎(主にウイルス性)、薬剤性髄膜炎、腫瘍関連髄膜炎などを十分に鑑別する。鑑別すべき疾患は、患者により異なり、鑑別不十分と考えられる際には専門医に速やかに相談すること。
全身性エリテマトーデス、全身性強皮症及び多発性筋炎/皮膚筋炎の3疾患にみられる臨床症候あるいは検査所見が同時にあるいは経過とともに混在して認められる。これらは一括して混合所見と呼ばれる。混合所見の中で頻度の高いものは、1) 多発関節炎、2) 白血球減少、3) 手指に限局した皮膚硬化、4) 筋力低下、5) 筋電図における筋原性異常所見、6) 間質性肺疾患、などである。なお、小児のMCTDは初発時には混合所見は乏しいことが多く、診断においては成人と異なった判断が必要である。
合併症としては、シェーグレン症候群 (25%)、慢性甲状腺炎 (10%)などである。
4.治療法
本症は自己免疫疾患であり、抗炎症療法と免疫抑制療法が治療の中心となる。NSAIDsもしばしば使用されるが、まれに無菌性髄膜炎が誘発される点に注意する。急性期には副腎皮質ステロイドや免疫抑制薬(シクロホスファミド、アザチオプリンなど)などの治療が中心となるが、一旦開始すると長期投与となるため、骨粗鬆症や糖尿病、感染症の誘発に注意する。全身性エリテマトーデス様の臨床症候が主体となる際には、抗マラリア薬(ヒドロキシクロロキン)の使用が推奨される。なお、中枢神経障害、急速に進行する肺障害・腎障害、血小板減少症を除いて大量副腎皮質ステロイドが必要になることは比較的少ない。
MCTDの生命予後を規定する肺動脈性肺高血圧症に対して、近年いくつかの選択的肺血管拡張薬が使用できるようになった。これらは肺血管拡張作用に加えて、肺動脈平滑筋細胞などの増殖を抑制する作用を有する。しかし、肺血管のリモデリングが進行した場合には、右心不全のコントロールがより重要になるため、循環器内科と共同して治療に当たる必要がある。労作時呼吸困難など症状が出現する前に診断・治療することが重要で、MCTD患者では定期的な心臓超音波検査の施行が推奨される。また、全身性強皮症や進行性線維化を伴う間質性肺疾患に対しては、臨床症候、呼吸機能検査、肺HRCT検査にて診断の上で、抗線維化薬であるニンテダニブが推奨される。
5.予後
発病からの5年生存率は96.9%で、初診時からの5年生存率は94.2%である。主死因は肺高血圧症、呼吸不全、心不全など心肺系の死因が全体の60%を占めている。
○ 要件の判定に必要な事項
1.患者数(令和元年度医療受給者証保持者数)
9,835人
2.発病の機構
不明(自己免疫性と考えられている。)
3.効果的な治療方法
未確立(根治的治療なし。)
4.長期の療養
必要(副腎皮質ステロイド長期投与)
5.診断基準
あり
6.重症度分類
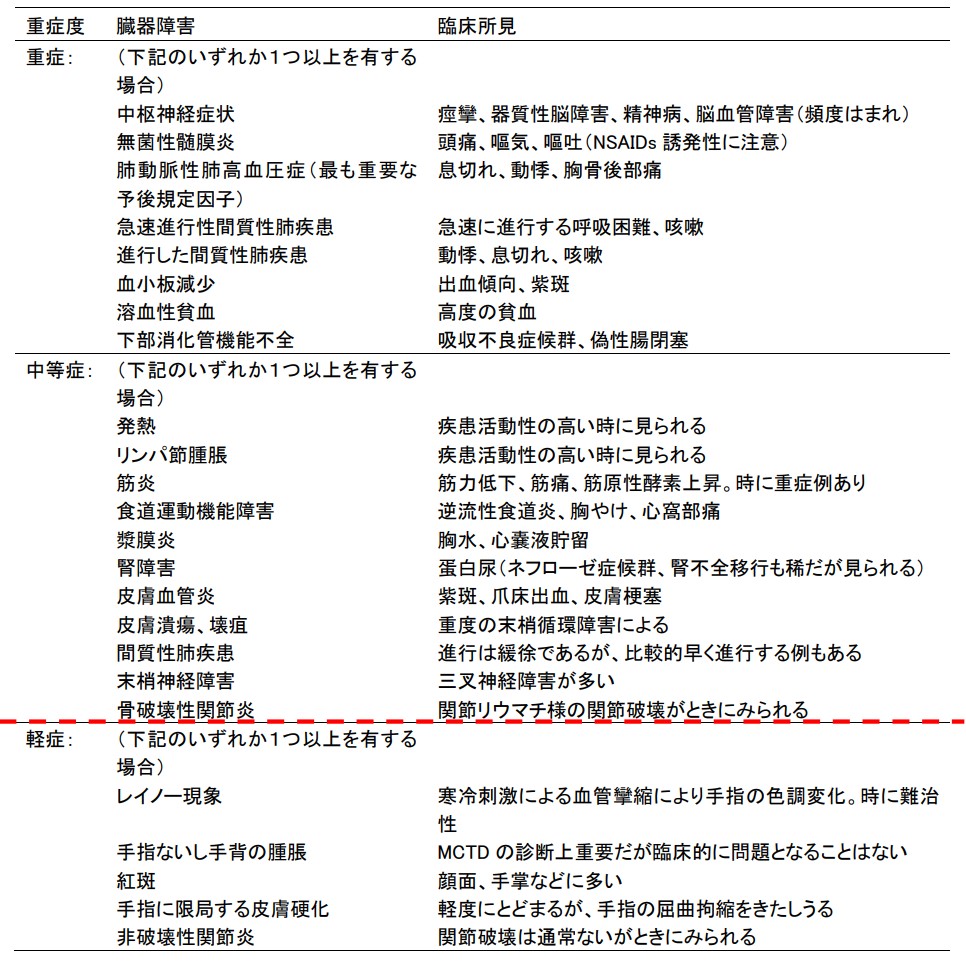
MCTDの障害臓器別の重症度分類を用いて中等症以上を対象とする。
○ 情報提供元
「自己免疫疾患に関する調査研究班」
混合性結合組織病(MCTD)分科会 産業医科大学医学部第1内科学講座 教授 田中良哉
<診断基準>
Definite1、2を対象とする。
1.共通所見
① レイノー現象 ② 手指ないし手背の腫脹
2.免疫学的所見
抗U1-RNP抗体陽性
・ 抗U1-RNP抗体の検出は二重免疫拡散法あるいは酵素免疫測定法(ELISA)のいずれでもよい。ただし、二重免疫拡散法が陽性で ELISA の結果と一致しない場合には、二重免疫拡散法を優先する。
※以下の予後、および臓器障害と関連する疾患標識抗体が陽性の場合は混合性結合組織病の診断は慎重に行う。
a. 抗二本鎖DNA抗体、抗Sm抗体
b. 抗トポイソメラーゼI抗体(抗Scl-70抗体)、抗RNAポリメラーゼⅢ抗体
c. 抗ARS抗体、抗MDA5抗体
3.特徴的な臓器所見
① 肺動脈性肺高血圧症 ② 無菌性髄膜炎 ③ 三叉神経障害
4.混合所見
(1) 全身性エリテマトーデス様所見
①多発関節炎
②リンパ節腫脹
③顔面紅斑
④心膜炎又は胸膜炎
⑤白血球減少(4,000/µL以下)又は血小板減少(10 万/µL以下)
(2)全身性強皮症様所見
①手指に限局した皮膚硬化
②間質性肺疾患
③食道蠕動低下又は拡張
(3)多発性筋炎/皮膚筋炎様所見
①筋力低下
②筋原性酵素上昇
③筋電図における筋原性異常所見
5.診断のカテゴリー
・Definite1:1の1所見以上が陽性,2が陽性、3の1 所見以上が陽性、以上3つをいずれも満たす場合。
・Definite2:1の1所見以上が陽性、2が陽性、4の(1)、(2)、(3)項より2項目以上からそれぞれ1所見以上が陽性、以上3つをいずれも満たす場合。
・小児(16歳未満の場合):1の1所見以上が陽性、2の所見が陽性、4の(1)、(2)、(3)項より1項目以上からそれぞれ1所見以上が陽性、以上3つをいずれも満たす場合。
<重症度分類>
MCTDの障害臓器別の重症度分類
中等症以上を対象とする。
※診断基準及び重症度分類の適応における留意事項
1.病名診断に用いる臨床症状、検査所見等に関して、診断基準上に特段の規定がない場合には、いずれの時期のものを用いても差し支えない(ただし、当該疾病の経過を示す臨床症状等であって、確認可能なものに限る。)。
2.治療開始後における重症度分類については、適切な医学的管理の下で治療が行われている状態であって、直近6か月間で最も悪い状態を医師が判断することとする。
3.なお、症状の程度が上記の重症度分類等で一定以上に該当しない者であるが、高額な医療を継続することが必要なものについては、医療費助成の対象とする。
治験を実施している指定難病を調べることができます。キーワード検索欄に指定難病名を入力し、検索してください。
治験・臨床研究情報検索サイト一覧
| 研究班名 | 自己免疫疾患に関する調査研究班 研究班名簿 |
|---|---|
| 情報更新日 | 令和6年4月(名簿更新:令和8年6月) |