22q11.2欠失症候群(指定難病203)
1. 22q11.2欠失症候群とは
22q11.2欠失症候群は、患者さんの80%に 先天性 疾患を合併し、精神発達遅延、特徴的顔貌や、胸腺低形成・無形成による免疫低下、口蓋裂・軟口蓋閉鎖不全、鼻声、低カルシウム血症を主徴とする症候群です。22番の染色体の微細欠失(顕微鏡では見えない程度の「22q11.2」と呼ばれる部分の欠失)が認められ、その部分に位置する約30個の遺伝子が欠失するため発症します。特にTBX1というヒトの心臓や大血管の形を決める遺伝子の欠失によって、心疾患としてファロー四徴症(+肺動脈弁欠損、肺動脈閉鎖、主要体肺側副動脈)や大動脈離断症の合併が多く見られます。
ファロー四徴症は、(1)左右の心室を分ける心室中隔という仕切りの壁の大きな穴(心室中隔欠損)、(2)全身へ血液を送る大動脈が左右の心室にまたがっている(大動脈騎乗)、(3)肺へ血液を送る肺動脈の右室の出口(漏斗部)が肺動脈弁と一緒に狭くなる(漏斗部狭窄および肺動脈狭窄)、(4)左右の心室の圧が等しくなり、右室が肥大する(右室肥大)の4つの特徴をもった心疾患です。
ファロー四徴症で肺動脈閉鎖をともなうものを、極型ファロー四徴症と呼ぶこともあります。22q11.2欠失症候群では、極型ファロー四徴症で主肺動脈が無く、主要体肺側副動脈によって肺への血流が供給される病型が多いのが特徴です。
ファロー四徴症についてはファロー四徴症(指定難病215)の病気の解説を参照してください。
2.この病気の患者さんはどのくらいいるのですか
4,000~5,000人に1人の頻度で発症します。ファロー四徴症の15%の患者さんが22q11.2欠失症候群です。
3.この病気はどのような人に多いですか
どのような人に多いかはわかっていません。
4.この病気の原因はわかっているのですか
染色体の微細欠失による遺伝子変異が原因です。
5.この病気は遺伝するのですか
ご本人の遺伝子変異は多くは突然変異ですが、家族性に認められることもあります。ご本人の遺伝子変異(22q11.2欠失)が子どもに遺伝する確率は50%です。
6.この病気ではどのような症状が見られますか
発達遅延、特徴的顔貌、先天性心血管疾患、粘膜下口蓋裂、胸腺低形成、低カルシウム血症などが多いですが、その他にも多様な臨床症状がでる可能性があります。
22q11.2欠失症候群で 生命予後 に深く関わるのが心血管疾患です。ファロー四徴症や大動脈弓離断の合併が多く認められます。ファロー四徴症およびその類縁疾患では、一般に肺血流が少なくなるので、チアノーゼ(低酸素血症のために口唇や爪床が紫色になる症状)が特徴的です。一部のファロー四徴症では、心不全症状(多呼吸、哺乳困難、体重増加不良)を呈することがあります。右室から肺動脈への流出路が、どの程度狭いかによって、チアノーゼの出現の時期と程度が変わります。生後2カ月以後には、この疾患に特有の「無酸素発作(チアノーゼ発作)」がみられることがあります。この発作は、最初は哺乳後、入浴後、よく寝た後に見られることが多く、重くなると一日中起きるようになります。症状は、急に不機嫌になり、チアノーゼと呼吸困難が強くなり、高度になると意識がなくなったり全身のけいれんを起こすことがあります。長時間続くと死亡することもあり、注意が必要です。チアノーゼが出現して6カ月以上経つと手足の指先が円く変形して、太鼓のばちのような形になります(ばち指と呼びます)。
大動脈弓離断では、新生児期から肺血流量増加による心不全症状がみられ、適切に診断されないと動脈管が閉鎖して下半身への血流が途絶え、ショックにより死亡することがあります(動脈管ショック)。
22q11.2欠失症候群では加えて、低身長、血小板減少、汎血球減少、けいれん、斜視、気管支軟化症、脳萎縮、白内障、尖足、側弯症、腎奇形、尿道下裂、鎖肛、鼠径ヘルニアなど多くの症状が合併する可能性があります。粘膜下口蓋裂による言葉の異常や精神発達遅延は、小学校入学前後から目立ってきます。学童期には学習障害が出て、特にストレスが強いと思春期に精神的な問題を抱えたり、その後精神的不調や精神疾患を発症することがあります。精神科など専門家への受診と治療が必要になります。
免疫力低下による 易感染性 は、ごく少数の症例以外ではあまり顕著ではなく、通常の予防接種が可能です。
7.この病気にはどのような治療法がありますか
染色体の異常そのものを治療する方法はありませんので、まず生命に直結する心臓病の治療が基本になります。新生児期からそれぞれの患者さんに適した心臓手術計画を立て、生涯にわたって、臨床症状に基づいた生活指導や治療を続ける必要があります。ファロー四徴症で高度肺動脈狭窄または肺動脈閉鎖の場合には、新生児期に短絡術が必要となることがあります。大動脈離断では、新生児期に大動脈の修復手術が必要となります。
低酸素血症を改善するための手術として、腕に血液を送る鎖骨下動脈という動脈と肺動脈をつなぐブラロックータウシッヒ短絡手術と、心室中隔欠損を閉鎖して狭い右室流出路を拡大形成する心内修復術があります。ブラロックータウシッヒ短絡手術は、新生児や乳児早期で体重が小さかったり、「無酸素発作」の改善のためや、肺動脈が細く左室が小さいために心内修復術をするのが難しい場合に行われます。ファロー四徴症の心内修復術は、通常生後1歳前後に行われることが多く、狭い右室流出路を広げるのに自分の肺動脈弁を残す方法(自己弁温存法)、右室流出路にパッチと呼ばれる膜を当てて拡大形成する方法(右室流出路パッチ拡大術)、人工血管などの導管を使って右室から肺動脈へ血流路を作製する方法(ラステリ手術)の3通りがあります(図1)。特に肺動脈閉鎖をともなう場合にはラステリ手術が行われます。
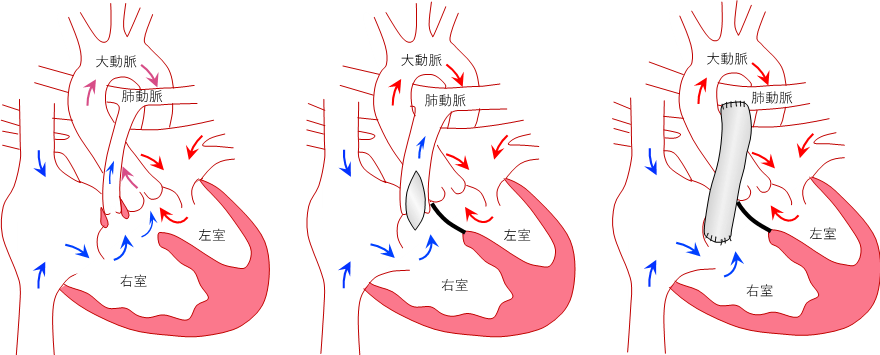
図1:左;ファロー四徴症、中央;右室流出路パッチ拡大術後、右;ラステリ手術後
中心肺動脈が無く主要肺体側副動脈の場合には、末梢肺動脈をまとめて左右の肺動脈をつくる手術(ユニフォーカリゼーション)が必要になることがあります。
小児期に心内修復術が行われた後、右室流出路が再び狭くなったり肺動脈が狭くなったりした場合には、バルーンカテーテルやステントで拡大する カテーテル治療 が行われる場合があります。また大動脈から肺動脈に異常なバイパス血管が出来た場合には、コイル塞栓術が行われる場合があります。
術後遠隔期、特に成人期には、肺動脈弁閉鎖不全の進行のために右心室が拡大し、右室の収縮が低下して 右心不全 を生じる場合があります。この場合には肺動脈弁置換術を必要とすることがあります。 成人期にラステリ手術後の導管に狭いところができて、導管を交換する手術が必要になることがあります。
8.この病気はどのような経過をたどるのですか
通常は、乳児期にチアノーゼを生じて、チアノーゼ発作や運動制限などのためにシャント手術や心内修復術が行われます。心内修復術を行えば、術後の状態にもよりますが、通常の日常生活は過ごせるようになることが多いです。しかし、激しい競技スポーツや運動は制限されることも多いです。加齢とともに肺動脈弁閉鎖不全が進行すると、運動時の息切れや日常生活の制限が起こるようになり、右室機能低下や三尖弁閉鎖不全を生じて心不全に陥ります。この場合、再手術(肺動脈弁置換術)を必要とすることがあります。また、肺動脈閉鎖でラステリ手術を行った場合、右室—肺動脈の導管の狭窄が進行することがあり、カテーテル治療による拡大を必要としたり、導管を交換する手術が必要になることがあります。
患者さんに精神発達遅延がある場合には、学校生活や就職などに際して社会支援が必要なことがあります。職業訓練などについても、学校や行政機関と相談して支援をしていく必要があります。
9.この病気は日常生活でどのような注意が必要ですか
手術前と術後の症状や状態によって異なります。激しい競技スポーツは制限されることがあります。運動時の準備運動/整理運動をきちんと行い、運動中の水分補給を心がけることが大切です。心臓病がある場合、抜歯や出血をともなう歯科治療、その他の外科手術時などには感染性心内膜炎の予防が必要です。術後の合併症が重くなければ妊娠出産は可能ですが、担当主治医および成人先天性心疾患専門医と事前によく相談して心機能を評価することが大切です。遺伝に関しては、主治医や遺伝カウンセラーに相談するとよいです。
10. 次の病名はこの病気の別名又はこの病気に含まれる、あるいは深く関連する病名です。 ただし、これらの病気(病名)であっても医療費助成の対象とならないこともありますので、主治医に相談してください。
ディジョージ症候群、円錐動脈幹異常顔貌症候群(高尾症候群)、口蓋帆・心臓・顔症候群
11.本病名の関連資料・リンク
① 小児・成育循環器学(改訂第2版). 日本小児循環器学会編集. 診断と治療社, 2024.
② 22q-pedia (東京大学22q研究事務局)
https://22q-pedia.net/about/
③ GRJ(GeneReviews Japan)22q11.2欠失症候群
https://grj.umin.jp/grj/22q11_2.htm
④ 先天性心疾患並びに小児期心疾患の診断検査と薬物療法ガイドライン(2018年改訂版)日本循環器学会.
https://www.j-circ.or.jp/cms/wp-content/uploads/2020/02/JCS2018_Yasukochi.pdf
⑤ 成人先天性心疾患診療ガイドライン(2025年改訂版)日本循環器学会.
https://www.j-circ.or.jp/cms/wp-content/uploads/2025/03/JCS2025_Yamagishi.pdf
⑥ 先天性心疾患術後遠隔期の管理・侵襲的治療に関するガイドライン(2022年改訂版)日本循環器学会.
https://www.j-circ.or.jp/cms/wp-content/uploads/2022/03/JCS2022_Ohuchi_Kawada.pdf
⑦ 心疾患患者の妊娠・出産の適応、管理に関するガイドライン(2018年改訂版)日本循環器学会.
https://www.j-circ.or.jp/cms/wp-content/uploads/2018/06/JCS2018_akagi_ikeda.pdf
治験を実施している指定難病を調べることができます。キーワード検索欄に指定難病名を入力し、検索してください。
治験・臨床研究情報検索サイト一覧
| 研究班名 | 先天性心疾患を主体とする小児期発症の心血管難治性疾患の救命率の向上、円滑な移行医療、成人期以降の予後改善を目指した総合的研究班 研究班名簿 |
|---|---|
| 情報更新日 | 令和7年12月(名簿更新:令和7年6月) |