肺胞蛋白症(自己免疫性又は先天性)(指定難病229)
○ 概要
1.概要
肺胞蛋白症(PAP)は1958年Rosenらにより記載され、我が国では1960年岡らによって紹介された稀少肺疾患である。肺胞蛋白症はサーファクタントの生成又は分解過程障害により肺胞腔内を主として末梢気腔内にサーファクタント由来物質である好酸性の顆粒状の蛋白様物質の異常貯留を来す疾患の総称である。このうち、指定難病は、自己免疫性PAPと先天性PAPである。
2.原因
自己免疫性PAPでは、顆粒球マクロファージコロニー刺激因子(GM-CSF)に対する中和自己抗体が存在し、肺胞マクロファージ、好中球の機能障害が病態に関与する。先天性PAPとしてはsurfactant protein (SP)-B、SP-C、ATP-binding cassette A3 (ABCA3)遺伝子の異常やGM-CSFレセプターの遺伝子変異が報告されているが(遺伝性PAP)、遺伝子異常の証明されていないものも少なくない。続発性PAPは骨髄異形成症候群などの血液疾患、粉塵やガスの吸入、感染症、リジン尿性蛋白不耐症、ベーチェット病等で認められる。
3.症状
自己免疫性PAPの男女比は2:1、診断時年齢の中央値は男女ともに51歳であった。症状は労作時呼吸困難(40%)、咳(10%)、喀痰、体重減少、発熱など。約30%の患者は無症状である。画像所見の割に症状が比較的軽微であることが本疾患の特徴である。先天性は重篤な場合が多い。続発性ではPAPの呼吸器症状に加えて原疾患の症状が加わる。
4.治療法
自己免疫性PAPには、洗浄療法(全肺洗浄あるいは区域洗浄)が行われる。試験的治療法としてGM-CSFの吸入療法が試みられる。先天性PAPは、対症療法等行うも予後は不良である。続発性PAPでは基礎疾患の治療、洗浄療法が行われるが効果は現時点では未確定である。肺移植が実施され移植肺にPAPが再発した報告がある。ステロイドの効果は一般に期待されない。
5.予後
自己免疫性肺胞蛋白症の5年生存率は96%、10年生存率は88%であるが、患者はこの間繰り返し全肺洗浄等の治療を要する場合が多い。先天性肺胞蛋白症の予後は極めて悪い。また続発性肺胞蛋白症は自己免疫性肺胞蛋白症に比べて予後は悪い。
○ 要件の判定に必要な事項
1. 患者数
約900人(自己免疫性PAP及び先天性PAP)
2. 発病の機構
不明(自己抗体が関与しているとされるが、発病機構の詳細は不明の点が多い。)
3. 効果的な治療方法
未確立(根治させる治療は未確立である。)
4. 長期の療養
必要(大多数の患者は繰り返し治療が必要である。)
5. 診断基準
あり(研究班作成の診断基準あり)
6. 重症度分類
重症度表を用いて管理区分重症度III以上を対象とする。
○ 情報提供元
「肺胞蛋白症、遺伝性間質性肺疾患に関する研究:重症難治化要因とその克服」
研究代表者 国立病院機構 近畿中央胸部疾患センター 臨床研究センター長 井上義一
<診断基準>
肺胞蛋白症(PAP)(自己免疫性、先天性)の診断基準
A.症状
症状は労作時呼吸困難(40%)、咳(10%)、喀痰、体重減少、発熱など。約30%の患者は無症状である。画像所見の割に症状が比較的軽微であることが本疾患の特徴である。続発性ではPAPの呼吸器症状に加えて原疾患の症状が加わる。先天性は重篤な場合が多い。
B.検査所見(以下の所見は診断の参考になる)
1.血液・生化学的検査所見
血清KL-6、サーファクタントプロテイン(SP)-A、SP-D、LDH高値
2.画像検査所見
高分解能CT(HRCT)にて、以下の所見を認める。
主要所見
1.すりガラス様陰影、通常両側性
2.小葉内間質肥厚像及び小葉間隔壁肥厚像
3.Crazy-paving pattern: 所見1と2の重なり合い
4.Consolidation
5.地図状分布 geographic distribution
6.Subpleural sparing
その他の所見
1.牽引性気管支拡張像
2.嚢胞
3.蜂巣肺
(PAPほぼ確実)Crazy-paving pattern(3.)が主体でこれに地図状分布(5.)、subpleural sparing(6.)が認められればHRCT診断でPAPがほぼ確実。
(PAP疑い)Crazy-paving patternのみを認めればPAP疑い。
(PAPを支持する所見)PAPほぼ確実とPAP疑いを「PAPを支持する所見」とする。
(鑑別を要するHRCTでCrazy-paving patternを呈する疾患)
ニューモシスチス肺炎、リポイド肺炎、ARDS、急性間質性肺炎、薬剤性肺炎、肺胞出血、細気管支肺胞上皮癌、非特異的間質性肺炎、器質化肺炎、サルコイドーシス、放射線肺炎、過敏性肺炎、肺静脈閉塞症、肺水腫、ウイルス性肺炎、レプトスピラ症、吸引性肺炎、肺胞微石症、菌状息肉症、カポシ肉腫など。
3.生理学的所見
自己免疫性PAP 患者は必ずしも呼吸機能上、換気障害を呈するわけではなく、画像的範囲、低酸素血症を加味した重症度を反映して、拘束性障害を呈してくる。肺活量の低下に比べ、低酸素血症や拡散能の低下をより生じやすい。
4.病理組織学的所見
PAP の基本的な所見:左右肺に肺病変を来たした症例で
(1)末梢気腔内に0.2 microns大の弱好酸性細顆粒状物質が充満する。細顆粒状物質に数十microns大の好酸性顆粒状物質が混在する。数microns大のlipid clefts が混在する
(2)末梢気腔内の細顆粒状物質はPAS 染色で陽性所見を示す。
(3)末梢気腔内の細顆粒状物質は免疫染色でSurfactant apoprotein A (SP-A)に陽性所見を示す。
PAP に伴うことがある所見:
(4)末梢気腔内に大型泡沫細胞が集積する。細胞質の崩壊過程を示す泡沫細胞を含む。
(5)肺胞領域の間質にリンパ球系細胞浸潤を見る。多くは軽度まで。
(6)間質性線維化病変が存在することがある。稀に線維化病変が著明な症例がある。
PAP の肺病変自体では陰性の所見(他疾患を鑑別すべき所見):
(1)腫瘍性病変
(2)肉芽腫性病変
(3)好中球あるいは好酸球の浸潤
(4)壊死病変
C.鑑別診断
以下の疾患を鑑別する。まれに合併する事もある。
ニューモシスチス肺炎、リポイド肺炎、ARDS、急性間質性肺炎、薬剤性肺炎、肺胞出血、細気管支肺胞上皮癌、器質化肺炎、サルコイドーシス、放射線肺炎、過敏性肺炎、肺水腫、吸引性肺炎、細菌性肺炎
骨髄異形成症候群などの血液疾患、粉塵やガスの吸入、感染症、リジン尿性蛋白不耐症、ベーチェット病等では続発性PAPを認める事がある。自己免疫性と続発性との鑑別のために必ず抗GM-CSF自己抗体の測定をすべきである。
<診断の流れ>
以下の表の基準でPAPの診断をまず行い、次に図に示すアルゴリズムに従いPAPの分類診断を行う。
表 PAPの診断基準
原則、以下の2項目を満たすこと
1.画像所見:胸部CT(原則、高分解能CT)撮影で、肺胞蛋白症を支持する所見を有する。
2.病理・細胞学的所見:下のa項又はb項を満たす。
a.気管支肺胞洗浄(BAL)液で白濁の外観を呈し、放置すると沈殿する。光顕で、パパニコロー染色でライトグリーンに染まる顆粒状の無構造物質の沈着と、泡沫状マクロファージ(foamy macrophage)がみられる。
b.病理組織(経気管支肺生検、外科的肺生検、剖検)で肺胞蛋白症を支持する所見がみられる。
注1)胸部高分解能CTにて、びまん性すりガラス様陰影(GGO)が見られる。GGOの分布は、自己免疫性肺胞蛋白症では地図状(辺縁が鮮明)であり、続発性肺胞蛋白症では均一(辺縁が不鮮明)であることが多い。上記画像所見参照。
注2)自己免疫性肺胞蛋白症の診断には血清中の抗GM-CSF自己抗体が陽性であることを必要とする。
抗GM-CSF自己抗体の測定がなされていない場合はこれまでの分類に従い特発性肺胞蛋白症と呼ぶに留める。
肺胞蛋白症の分類診断のアルゴリズム
以下のアルゴリズムに従って自己免疫性PAP、続発性PAP、先天性PAP(遺伝子解析がなされた場合遺伝性PAPと呼び先天性PAPに含める)、未分類PAPを診断し、自己免疫性PAP及び先天性PAP(遺伝性PAP)を指定難病の対象とする。
図 肺胞蛋白症診断のアルゴリズム
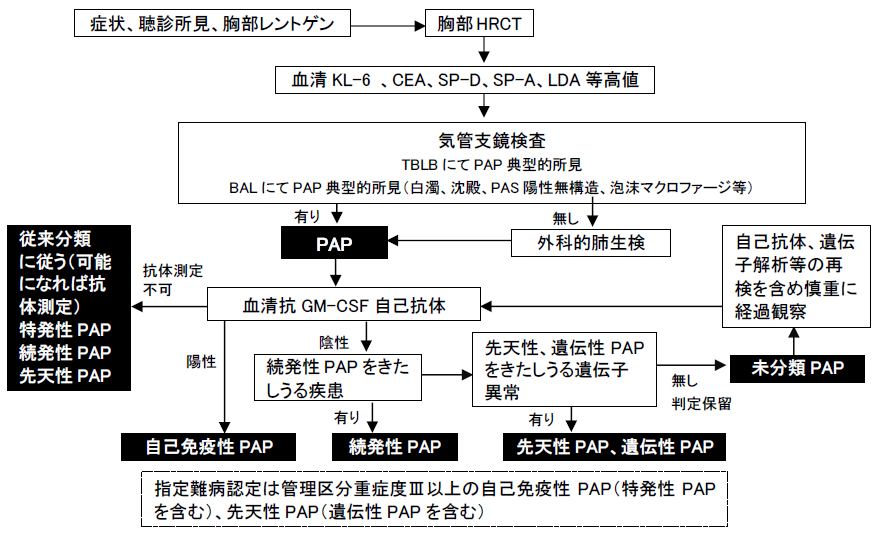
注: 続発性PAPを来しうる疾患の一覧(平成28年研究班成績)。尚、じん肺等に伴うPAPは歴史的に続発性と呼ばれていたが、多くは抗GM-CSF抗体陽性である。抗GM-CSF抗体による診断が推奨される。
|
疾患名 |
研究班成績(n=74) |
|
|
Hematological disorders |
65 (88%) |
|
|
|
Myelodysplastic syndrome |
50 (68%) |
|
|
Chronic myeloid leukemia |
3 (4%) |
|
|
Myelofibrosis |
2 (3%) |
|
|
Acute myeloid leukemia |
3 (4%) |
|
|
Acute lymphoid leukemia |
1 |
|
|
Adult T cell leukemia |
2 (3%) |
|
|
Aplastic anemia |
2 (3%) |
|
|
Lymphoma |
1 |
|
|
Multiple myeloma |
1 |
|
|
Plasmacytoma |
0 |
|
|
Alymphoplasia |
0 |
|
|
Essential thrombocytosis |
0 |
|
Infectious diseases |
2 (3%) |
|
|
Autoimmune diseases |
5 (7%) |
|
|
Post organ transplantation |
0 |
|
|
Non hematological malignancy |
0 |
|
<重症度分類>
【肺胞蛋白症の重症度と管理区分重症度】
PAP重症度に難治例等を考慮し管理区分重症度を計算する。管理区分重症度III以上を対象とする。
【PAP重症度】
|
重症度(DSS) |
症状 |
PaO2 |
|
1 |
無し |
PaO2≧70 Torr |
|
2 |
有り |
PaO2≧70 Torr |
|
3 |
不問 |
70 Torr>PaO2≧60 Torr |
|
4 |
不問 |
60 Torr>PaO2≧50 Torr |
|
5 |
不問 |
50 Torr>PaO2 |
【管理区分重症度】
以下の場合、難治例として、重症度を1度加えて管理区分重症度とする(I~VIで表記)。その場合、管理区分重症度の後に( )を附記し詳細を記入。 例 管理区分重症度: III (肺線維症合併)
(1)明らかな肺線維症の合併
(2)反復、継続する感染症合併
(3)CPAPの場合
(4)6分間歩行試験で、Sp0290%未満を認める場合
※診断基準及び重症度分類の適応における留意事項
1.病名診断に用いる臨床症状、検査所見等に関して、診断基準上に特段の規定がない場合には、いずれの時期のものを用いても差し支えない(ただし、当該疾病の経過を示す臨床症状等であって、確認可能なものに限る。)。
2.治療開始後における重症度分類については、適切な医学的管理の下で治療が行われている状態であって、直近6か月間で最も悪い状態を医師が判断することとする。
3.なお、症状の程度が上記の重症度分類等で一定以上に該当しない者であるが、高額な医療を継続することが必要なものについては、医療費助成の対象とする。
- 井上義一、中田光監修. 肺胞蛋白症の診断、治療、管理の指針. 肺胞蛋白症の難治化要因の解明と診断、治療、管理の標準化と指針の確立研究班.平成24年3月.
- Kiramura T, et al. Idiopathic pulmonary alveolar proteinosis as an autoimmune disease with neutralizing antibody against granulocyte/macrophage colony-stimulating factor. J Exp Med. 1999; 190: 875-80.
- Inoue Y, et.al. Characteristics of Large Cohort of Patients with Autoimmune Pulmonary Alveolar Proteinosis in Japan. Am J Respir Crit Care Med. 2008; 177: 752-762.
- Sakagami T, et al.. Patient-derived granulocyte/macrophage colony-stimulating factor autoantibodies reproduce pulmonary alveolar proteinosis in nonhuman primates. Am J Respir Crit Care Med. 2010; 182: 49-61.
- Ishii H, et al. Clinical features of secondary pulmonary alveolar proteinosis: pre-mortem cases in Japan. Eur Respir J. 2011 Feb;37(2):465-8
- Suzuki T, et al. Pulmonary macrophage transplantation therapy. Nature. 2014 Oct 23;514(7523):450-4
- Kumar A, et al.. Pulmonary Alveolar Proteinosis in Adults: Pathophysiology and Clinical Approach. Lancet Respir Med. 2018 Jul;6(7):554-565.
- Trapnell BC, et al.. Pulmonary Alveolar Proteinosis. Nat Rev Dis Primers. 2019 Mar 7;5(1):16.
- Tazawa R, et al. Inhaled GM-CSF for Pulmonary Alveolar Proteinosis. N Engl J Med. 2019 Sep 5;381(10):923-932
- Trapnell BC, et al. Inhaled Molgramostim Therapy in Autoimmune Pulmonary Alveolar Proteinosis. Engl J Med. 2020 Oct 22;383(17):1635-1644
- Sakaue S et al. Genetic determinants of risk inautoimmune pulmonary alveolar proteinosis. Nat Commum. 2021 Feb 15;12(1):1032
- 日本肺胞蛋白症患者会
https://pap-net.jp
治験を実施している指定難病を調べることができます。キーワード検索欄に指定難病名を入力し、検索してください。
治験・臨床研究情報検索サイト一覧
| 研究班名 | びまん性肺疾患に関する調査研究班 研究班名簿 |
|---|---|
| 情報更新日 | 令和5年10月(名簿更新:令和8年6月) |