脳内鉄沈着神経変性症(指定難病121)
○ 概要
1.概要
脳内鉄沈着神経変性症は、 OMIMのNBIA:Neurodegeneration with brain iron accumulationシリーズとされてる6疾患とその他の4疾患の現時点では合計10疾患の総称で、中枢神経系に鉄が沈着し、脳機能障害を呈する疾患群である。鉄の沈着部位により若干症状に差異はあるが一般に進行性の精神・神経症状と随意運動障害、ジストニアを中心とする不随意運動を中核症状とするものが多い。発症年齢は多くは小児期であるが、成人発症の病型もある。
一般に、予後は不良であり、原因療法はなく対症療法に留まる。一部の疾患では脳深部刺激療法もなされ、症状の緩和が報告されている。
2.原因
脳内鉄沈着神経変性症では全ての病因遺伝子が同定されている。多くは常染色体潜性遺伝(劣性遺伝)様式で小児期発症であるが、神経フェリチン症は常染色体顕性遺伝(優性遺伝)様式で成人期発症、成人期における小児期発症の神経変性を伴う停滞性脳症(SENDA)もしくはβ-プロペラ足場タンパク変異に伴う神経変性症(BPAN):NBIA5はX連鎖性顕性遺伝(優性遺伝)様式で20-30歳代に発症することが多い。原因遺伝子が同定され、遺伝子産物が既知となっていても必ずしも病態のすべての原因とならないことも多いため、脳内鉄沈着神経変性症の疾患名は○○に伴う神経変性症と命名されている(例:pantothenate kinase-associated neurodegeneration)。
3.症状
脳内鉄沈着神経変性症の臨床症状は精神症状として精神運動発達遅滞、退行、認知障害、神経症状としては歩行障害などの随意運動障害、ジストニアを中心とする不随意運動の発現を中核症状とする。発症年齢は多くは小児期であるが、成人発症の病型もある。病型により特徴的な症状として、乳児神経軸索ジストロフィー症(もしくはPLA2G6変異に伴う神経変性症(PLAN)、NBIA2a、 2b)では小脳運動失調症状、神経フェリチン症(NBIA3)では舞踏運動などがある。遺伝性ジストニアと同様に、同一遺伝子変異による病型であっても、家系間、家系内で病像が異なることがあり、留意すべきである。検査所見としては、脳内鉄沈着神経変性症すべての病型で、MRIで基底核への鉄沈着像を認める。その他検査所見では、無セルロプラスミン血症(aceruloplasminemia)ではセルロプラスミン欠損、糖尿病などを、神経フェリチン症(neuroferritinopathy)ではフェリチン値低値を認める。脳内鉄沈着神経変性症NBIAに属する疾患の各病型の特徴と画像所見の詳細については各論で付記する。
4.治療法
根治療法は確立されていない。対症療法としては、薬物治療(抗コリン剤、抗てんかん薬、L-dopa製剤など)やボツリヌス毒素の局部注射療法、定位脳手術(後腹側淡蒼球凝固術、視床凝固術、脳深部刺激療法)がある。
5.予後
脳内鉄沈着神経変性症は緩徐進行性の病像を示し、予後不良である。病状の進行の程度は介入方法の有無、介入時期によって様々であり、長期的な医療の介入が必要である。
○ 要件の判定に必要な事項
1. 患者数
約100人未満
2. 発病の機構
病理変化および大脳基底核への鉄の沈着が共通要素であるため、金属代謝を含めた病態解明と共に、病因遺伝子の機能から推定される発症病理に対応する治療薬の開発が進捗しつつある。
3. 効果的な治療方法
未確立(対症治療は存在するが、根治療法は未確立。)
4. 長期の療養
必要(症状が継続し、進行性の経過をたどる。)
5. 診断基準
あり(研究班作成の診断基準)
6. 重症度分類
Barthel Indexを用いて、85点以下を対象とする。
○ 情報提供元
「神経変性疾患領域における基盤的調査研究班」
研究代表者 国立病院機構松江医療センター 中島健二
「Pantothenate kinase-associated neurodegeneration(PKAN)の診断基準作成と実態調査に関する研究班」
研究代表者 国立病院機構相模原病院 神経内科学 医長 長谷川一子
「神経フェリチン症の診断基準の構築と調査に関する研究班」
研究代表者 埼玉医科大学国際医療センター 神経内科・脳卒中内科 教授 高尾昌樹
<診断基準>
いずれかの病型で臨床所見、遺伝子診断によりDefinite(確定診断)とされたものを対象とする。
脳内鉄沈着神経変性症は病型により臨床症状が異なるため、共通する事項と各病型の診断基準については以下に別に記載する。
遺伝子診断を施行する際は日本神経学会「神経疾患の遺伝子診断に関するガイドライン」も参考とすること。
脳内鉄沈着神経変性症に共通する事項を以下に示す。
1.診断のカテゴリー
Definite:いずれかの病型に合致する2.臨床症状または3.特記すべき検査所見にかかげる内容を1つ以上認め、かつ鑑別診断を除外し遺伝子診断により確定診断されたもの
Probable:いずれかの病型に合致する2.臨床症状または3.特記すべき検査所見にかかげる内容を1つ以上認め、かつ鑑別診断を除外するが遺伝子診断が未確定のもの
2.鑑別診断(脳内鉄沈着神経変性症の全ての病型において鑑別すべき疾患。)
(1) 代謝性疾患:ウィルソン病、メンケス病、αfucosidosis、Glutaric aciduria I、リー脳症
(2) 遺伝性神経変性疾患:神経セロイドリポフスチン症(Neuronal ceroid lipofuscinosis)、ハンチントン病、神経有棘赤血球症
(3) リピドーシス:β-hexosaminidase A欠損症、GM1-galactosidase欠損症、ニーマン・ピック病
(4) 小児期発症の遺伝性小脳性運動失調 (SCA1、2、3、17など)、歯状核赤核・淡蒼球ルイ体萎縮症(DRPLA)、家族性痙性対麻痺30型、遺伝性感覚性ニューロパチーⅡC型(HSNⅡC)
(5) 遺伝性パーキンソニズム:PARK5、6、15など
(6) 遺伝性ジストニアに属する疾患群
(7) 脳血管障害
(8) 抗精神病薬などによる遅発性ジストニア
(9)レット症候群
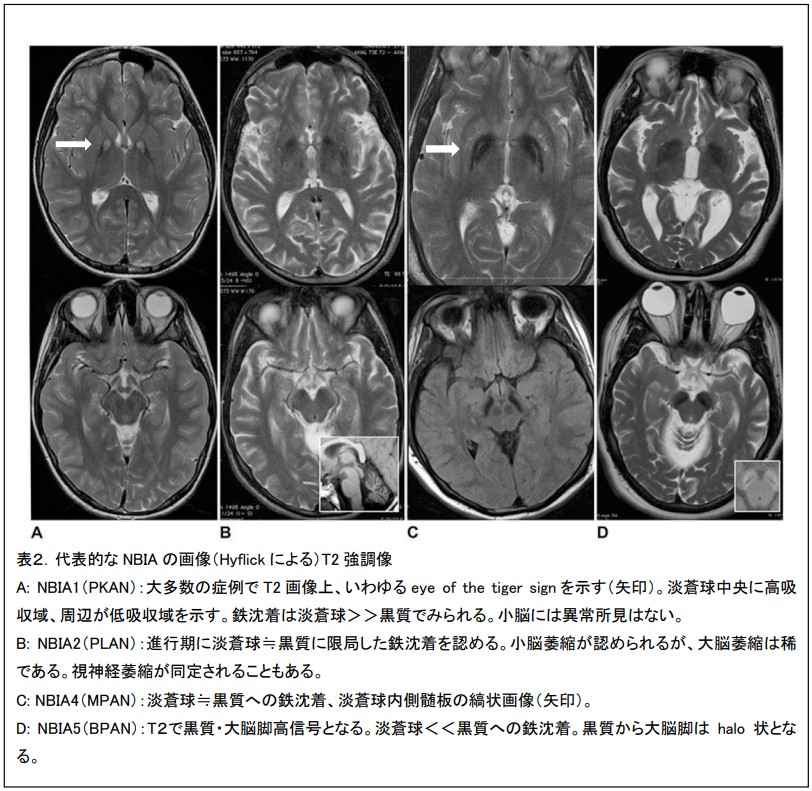
各病型の診断基準について以下に示す。
<パントテン酸キナーゼ変異を伴う神経変性症Pantothenate kinase-associated neurodegeneration:PKAN、別名NBIA 1>
1.病因遺伝子と概要
パントテン酸キナーゼ2pantothenate kinase2:PANK2遺伝子変異による疾患で、ジストニアを主症状とする。脳内鉄沈着神経変性症の代表的疾患である。全経過は古典型で15年程度、非典型で15~40年とされる。PANK2はCoenzyme A生合成関連疾患の律速段階に当たる酵素である。
(1)遺伝様式:常染色体潜性遺伝(劣性遺伝)(遺伝子座 22q13、遺伝子PKAN2、 遺伝子産物 PKAN(Pantothenate kinase 2)、MIM ID #234200、 *606157)。
(2)発症年齢:古典型:小児期(多くは6歳以下)、非典型:小児期~青年期(多くは10歳以上)
(3)頻度:1~3/1,000,000
2.臨床症状
(1)古典型:
75%の症例は古典型とされる。歩行障害、姿勢障害、巧緻障害で発症し、錐体外路症状、発達障害が加わる。錐体外路症状の多くはジストニアで、筋強剛や舞踏運動がそれに続く。ジストニアの初発症状は下肢であるが、脳神経領域、四肢に見られる。口部ジストニアにより咬舌を来すこともある。錐体路症状も通常見られる。発症早期に網膜色素変性症、視神経萎縮が2/3の症例で合併する。症状は進行性で発症から10~15年で歩行不能となることが多い。てんかん発作はまれである。
(2)非典型:
25%を占める非典型の症例では、発話障害や精神症状が目立ち、より緩徐に進行する。発話障害としては40%の症例で反復言語か構音障害が多い。その後ジストニアをみるが、古典型よりも程度は軽症で、15~40年程度で歩行不能となることが多い。すくみ足の頻度も高い。約1/3の症例で精神症状(衝動性障害、強迫性障害が多い)か前頭側頭葉型認知症が見られる。症例によっては運動症状が明らかでなく、精神症状で推移する場合もある。網膜色素変性症は通常合併しない。発症年齢が高くなるにつれ、パーキンソニズムが目立つ傾向となり、若年性パーキンソン病として治療されている症例もみられる。
(3)その他:
中間表現型と呼ばれる10歳代以前に発症するが進行が遅い型、10歳代に発症し進行が速く20歳代に歩行不能となる例などがある。その他Tourette症候群、純粋アキネジア、運動ニューロン疾患類似の病態を呈する症例、若年発症のパーキンソニズムを示す症例などが報告されている。
HARP症候群(hypoprebetalipoproteinemia、acanthocytosis、retinitis pigmentosa、pallidal degeneration、OMIM 607236)も遺伝子変異がPANK2に見られたことからPKANに包含された。
3.特記すべき検査所見
(1)脳MRI画像: eye-of-the tiger signを認める。これは1.5テスラ以上のMRIのT2強調画像で、強い低輝度を示す淡蒼球内の内側領域に高輝度を認めることを指す。特異度の高い所見で有り、MRI所見からPKANの診断に至る例が少なくないが、擬陽性例、陰性例もあることに留意する必要がある。画像でeye-of-the tiger signに類似の画像を呈する疾患としては後述するNBIA3、 NBIA4、CO中毒等が報告されている。
(2)網膜電図:網膜障害パターンを示す。
(3)一般検査所見:8%の症例で有棘赤血球症を示す。
4.遺伝子診断
Pantothenate kinase 2遺伝子PANK2の病的変異を認める。
5.鑑別疾患
ウィルソン病、メンケス病、αfucosidosis、Glutaric aciduria I、リー脳症、神経セロイドリポフスチン症(Neuronal ceroid lipofuscinosis)、ハンチントン病、神経有棘赤血球症、β-hexosaminidase A欠損症、GM1-galactosidase欠損症、ニーマン・ピック病、小児期発症の遺伝性小脳性運動失調、歯状核赤核・淡蒼球ルイ体萎縮症(DRPLA)、家族性痙性対麻痺30型、遺伝性感覚性ニューロパチーⅡC型(HSNⅡC)、遺伝性パーキンソニズム、遺伝性ジストニアに属する疾患群、脳血管障害、抗精神病薬などによる遅発性ジストニア、レット症候群。
<乳児神経軸索ジストロフィー(Infantile neuroaxonal dystrophy:INAD)、NBIA2、別名PLA2G6変異に伴う神経変性症:calcium -independent phospholipase A2 group VI (PLA2G6) associated neurodegeneration: PLAN、国際運動障害学会の表記ではNBIA/DYT/PARK-PLA2G6>
1.病因遺伝子と概要
脂質代謝関連酵素由来の脳内鉄沈着神経変性症で、病理学的に中枢神経系と末梢神経型にスフェロイドがみられ、進行性に精神運動退行を示す。生前診断が可能。症例数が少ないため全経過は明確ではないがNBIA2Aで平均9.4年、NBIA2Bで約20年とされる。
(1)遺伝様式:常染色体潜性遺伝(劣性遺伝)(遺伝子座 22q13.1、原因遺伝子 PLA2G6(phospholipase A2、group VI)がINADの79%の症例で同定された。ミスセンス変異が大半を占める。MIM ID #256600、#610217、#603604)
(2)発症年齢:古典型:乳児期~幼児期(平均1歳歳)、非典型:小児期(平均4.4歳歳)
(3)頻度:1/1,000,000
2.臨床症状
進行性の精神症状、低緊張、深部腱反射亢進、四肢麻痺を示す。
(1)古典型:NBIA2A
精神運動退行と体幹の低緊張、深部腱反射亢進、進行性の四肢麻痺を生後6か月から3年の間に示す。多くの症例では次第に痙性四肢麻痺となるが、1/3の症例では反射消失性の脱力のままである。全例でジストニア、痙縮、球症状、小脳症状を認める。発症後5年くらいまで歩行可能であることが多い。約半数で失調性のあるいは他の要因による歩行障害を示し、視神経症状(視神経萎縮、斜視、眼振など)を認める。1/3の症例ではてんかん発作を認める。平均死亡年齢は9.4歳である。
(2)非典型:NBIA2B
発症時期は古典型より遅いが多くは10歳前に発症。主症状は不安定歩行、失調性歩行障害である。言語発達は遅れ、社会的な意思疎通はできない。視神経萎縮、眼振、痙攣発作は古典型と同様であるが、体幹の低緊張は見られない。
(3)Karak症候群
臨床像として早期発症小脳失調、ジストニア、痙縮、知能低下があり、MRIで小脳萎縮、淡蒼球と黒質に鉄沈着を認める症候群として報告されたが、PLA2G6遺伝子変異(複合ヘテロ接合体)が同定され、脳内鉄沈着神経変性症の一つとされている。
3.特記すべき検査所見
(1)電気生理学的検査:脱神経所見、神経伝導速度低下は1/3に認める。脳波検査で速波を認める。
(2)MRI画像検査:95%の症例で小脳萎縮、50%の症例で淡蒼球、黒質に鉄の沈着を認める。小脳のグリオーシスに対応してT2強調画像で小脳の高輝度を認める。また、脳梁、大脳白質で異常を認める頻度が高い。非典型:NBIA2BのMRI像では小脳萎縮は83%の症例に留まり、淡蒼球や黒質の鉄沈着が目立つ。
(3)末梢神経生検:PLA2G6遺伝子変異陽性症例の87%で、末梢神経生検組織において軸索スフェロイドを認める。
4.遺伝子診断
PLA2G6の病的変異を認める。
5.鑑別疾患
ウィルソン病、メンケス病、αfucosidosis、Glutaric aciduria I、リー脳症、神経セロイドリポフスチン症(Neuronal ceroid lipofuscinosis)、ハンチントン病、神経有棘赤血球症、β-hexosaminidase A欠損症、GM1-galactosidase欠損症、ニーマン・ピック病、小児期発症の遺伝性小脳性運動失調、歯状核赤核・淡蒼球ルイ体萎縮症(DRPLA)、家族性痙性対麻痺30型、遺伝性感覚性ニューロパチーⅡC型(HSNⅡC)、遺伝性パーキンソニズム、遺伝性ジストニアに属する疾患群、脳血管障害、抗精神病薬などによる遅発性ジストニア、レット症候群。
<神経フェリチン症(Neuroferritinopathy:FTL、別名NBIA3)>
1.病因遺伝子と概要
脳内鉄沈着神経変性症の中で唯一鉄に直接関連する疾患である。神経フェリチン症は、フェリチン軽鎖遺伝子FTL変異に関連する多彩な神経症候を数十年にわたり認める疾患である。病理学的には変異フェリチン軽鎖、正常フェリチン軽鎖および重鎖が、神経細胞体及び核内、グリア細胞核内に蓄積する。
(1)遺伝様式:常染色体顕性遺伝(優性遺伝)(遺伝子座19q13.3-q13.4、原因遺伝子FTL gene の第4exonの460insA変異、まれに458dupA、遺伝子産物 FTL:ferritin light chain MIM ID #606159、#134790)
(2)発症年齢:成人期(平均39歳)、小児期発症はまれである。
(3)頻度:不明
2.臨床症状
成人発症の舞踏運動、またはジストニアを1~2肢に認め、軽度の認知機能障害を伴う。錐体外路症は舞踏運動が50%、局所性ジストニア43%、パーキンソニズム7.5%で、急性バリスムや顔面痙攣、書痙はまれである。口下顎ジストニアや発声困難が見られる。顔面のジストニアは動作特異的で会話の際に広頸筋や前頭筋が収縮する、他の脳神経には問題ない。進行すると舞踏運動とジストニア双方が見られるようになる。5~10年で他肢に広がり、発症後20年くらいで全身性となるが、非対称性である。小脳失調、動作性振戦、認知症は目立たないことが多い。
458dupAは進行が速く、パーキンソニズムが目立ち、認知障害、小脳失調が見られる(460insAとの差異)。日本人家系(c.469_484dup16nt)も報告され10歳代に手指の振戦が見られ、低緊張性で、発声困難、小字症、歩行障害を示す。振戦は姿勢時が主体で、神経フェリチン症(neuroferritinopathy)で見られる典型的な錐体外路症状を示さない。
3.特記すべき検査所見
(1)臨床検査:血清フェリチン濃度はほとんどの男性、更年期以降の女性で低下する。更年期以前の女性では血清フェリチン濃度の低下は1/4に留まる。
(2)脳MRI画像:発症早期には赤核、尾状核、淡蒼球、被殻、視床、黒質、大脳皮質がT2強調画像で低輝度となる。進行期となり組織障害が増悪すると、淡蒼球、尾状核が高輝度となる。これはおそらく変性による浮腫、嚢胞と思われる。C468_484dup 16int症例では小脳萎縮を認める。T2*による撮像の報告もあり、最早期や発症前症例で淡蒼球や黒質が低輝度となる。
4.遺伝子診断
FTL遺伝子の病的変異を認める。
5.鑑別診断
ウィルソン病、メンケス病、αfucosidosis、Glutaric aciduria I、リー脳症、神経セロイドリポフスチン症(Neuronal ceroid lipofuscinosis)、ハンチントン病、神経有棘赤血球症、β-hexosaminidase A欠損症、GM1-galactosidase欠損症、ニーマン・ピック病、小児期発症の遺伝性小脳性運動失調、歯状核赤核・淡蒼球ルイ体萎縮症(DRPLA)、家族性痙性対麻痺30型、遺伝性感覚性ニューロパチーⅡC型(HSNⅡC)、遺伝性パーキンソニズム、遺伝性ジストニアに属する疾患群、脳血管障害、抗精神病薬などによる遅発性ジストニア、レット症候群。
<ミトコンドリア膜タンパク質に伴う神経変性症mitochondrial membrane protein-associated neurodegeneration:MPAN、別名NBIA4>
1.病因遺伝子と概要
脂質代謝関連の脳内鉄沈着神経変性症で、ミトコンドリア膜に局在するC19orf12遺伝子変異により発症する疾患で、痙性麻痺で発症し、下位運動ニューロン障害、視神経萎縮、認知症、精神症状を特徴とする。罹病期間は5~10年である。
(1)遺伝様式:常染色体潜性遺伝(劣性遺伝)(遺伝子座;19q12遺伝子変異;C19orf12、 遺伝子産物;C19orf12、 OMIM #624298)
(2)発症年齢:小児期(平均9歳)
(3)頻度:不明
2.臨床症状
臨床像は進行性の認知障害、行動障害が目立つ。四肢のジストニア、痙直(進行性痙性対(四肢)麻痺)、パーキンソニズム、構音障害、小脳失調、視神経萎縮、網膜色素変性症を認める。精神症状としては学習障害、知的退行を認める。緩徐進行を示す。自律神経も障害され腸管蠕動異常や排尿障害を認め、便、尿失禁を早期から認めることが多い。小児期発症例では下位運動ニューロン障害も生じ、筋萎縮、深部腱反射消失を来す。高齢発症例では精神症状が目立つことがある。
3.特記すべき検査所見
(1) 筋電図検査:軸索性運動ニューロパシーを示し早期には上位運動ニューロパシー、進行すると下位運動ニューロパシーが加わる。小児期発症例では下位運動ニューロン症状が目立ち、運動ニューロン病疾患MNDと誤診されることもある。
(2) 脳MRI画像:T2強調像では発症早期から淡蒼球と黒質への淡明化(鉄沈着)と大脳、小脳の萎縮を認める。
4.遺伝子診断
C19orf12遺伝子の病的変異を認める。
5.鑑別診断
ウィルソン病、メンケス病、αfucosidosis、Glutaric aciduria I、リー脳症、神経セロイドリポフスチン症(Neuronal ceroid lipofuscinosis)、ハンチントン病、神経有棘赤血球症、β-hexosaminidase A欠損症、GM1-galactosidase欠損症、ニーマン・ピック病、小児期発症の遺伝性小脳性運動失調、歯状核赤核・淡蒼球ルイ体萎縮症(DRPLA)、家族性痙性対麻痺30型、遺伝性感覚性ニューロパチーⅡC型(HSNⅡC)、遺伝性パーキンソニズム、遺伝性ジストニアに属する疾患群、脳血管障害、抗精神病薬などによる遅発性ジストニア、レット症候群。
<成人期神経変性を伴う小児期非進行性脳症static encephalopathy of childhood with neurodegeneration in adulthood: SENDA/ベータプロペラタンパク質に関連した神経変性症beta-propeller protein-associated neurodegeneration: BPAN/NBIA5>
1.病因遺伝子と概要
オートファジー関連脳内鉄沈着神経変性症で、比較的重度の知的障害と軽度の運動発達遅滞で小児期に発症する。成人期以降に認知症やジストニア・パーキンソニズムが急速に進行し、数年の経過で臥床状態となる。
(1)遺伝様式:X染色体顕性遺伝(優性遺伝)様式(遺伝子座;Xp11.23、病因遺伝子;WDR45、遺伝子産物;WD repeat domain phosphoinositide interacting protein 4:WIPI4、原則de novo変異である。X染色体の不活性化により、正常異常アレルの発現の偏りが発症に関与する。男性は一般的には胎生致死となる。OMIM#300894)
(2)発症年齢:小児期~成人期
(3)頻度:不明
2.臨床症状
小児期に知的障害運動発達遅滞で発症し、成人期以降に進行性の運動・認知機能低下、錐体路症状、睡眠障害を示す。発語は数単語に留まる。聴覚認知機能の障害は言語に比較して軽度で、簡単な指示理解は可能である。小児期に多彩なてんかん症状を示すことが少なくないが、成人期以降は発作頻度は軽減する。遺伝子が同定された際にstatic encephalopathy with neurodegeneration in childhood: SENDAと称された。軽症例では手指の常同運動が目立ち、当初、非典型Rett症候群とされた症例もある。成人期になるとジストニア・パーキンソニズム、歩行障害(小股歩行やfreezing)、認知症を示し、認知症は比較的急に悪化する。パーキンソニズムはL-dopaに反応するが、短期間で症状の日内変動やジスキネジアなどを示す。症状の強弱はX染色体不活化の偏りの程度、遺伝子変異部位により異なる可能性などが想定されているが、結論はでていない。 殆どの症例は女性であり、男性症例は致死的である。
3.特記すべき検査所見
(1)脳MRI画像:鉄沈着は淡蒼球と黒質にみられ、他のNBIAと異なり黒質>淡蒼球である。T1で黒質~大脳脚にかけてhalo状となる。脳梁は非薄化し、大脳と小脳は萎縮性である。
(2)神経病理像:淡蒼球と黒質に鉄沈着と神経細胞脱落を認める。synucleinopathy像はなく、進行すると大脳にびまん性にtauopathy像が見られる。軸索スフェロイドを黒質、淡蒼球、延髄、橋、視床に認める。
一般検査所見に特異的所見はない。
4.遺伝子診断
WDR45遺伝子の病的変異を認める。
5.鑑別疾患
ウィルソン病、メンケス病、αfucosidosis、Glutaric aciduria I、リー脳症、神経セロイドリポフスチン症(Neuronal ceroid lipofuscinosis)、ハンチントン病、神経有棘赤血球症、β-hexosaminidase A欠損症、GM1-galactosidase欠損症、ニーマン・ピック病、小児期発症の遺伝性小脳性運動失調、歯状核赤核・淡蒼球ルイ体萎縮症(DRPLA)、家族性痙性対麻痺30型、遺伝性感覚性ニューロパチーⅡC型(HSNⅡC)、遺伝性パーキンソニズム、遺伝性ジストニアに属する疾患群、脳血管障害、抗精神病薬などによる遅発性ジストニア、レット症候群。
<コエンザイムA合成酵素タンパク質関連神経変性症Coenzyme A synthase (COASY) protein-associated Neurodegeneration:CoPAN、別名NBIA6>
1.病因遺伝子と概要
CoA生合成に関連した脳内鉄沈着神経変性症で、NBIA1:PKANと臨床像が類似する。小児期発症の歩行障害、認知機能障害を伴うジストニア痙縮を示す。緩徐進行性である。
(1)遺伝様式;常染色体潜性遺伝(劣性遺伝)様式(遺伝子座;17q21.2、病因遺伝子COASY、遺伝子産物はCoenzyme A synthase、 OMIM#615643、#615643)
(2)発症年齢;小児期(平均2歳)
(3)頻度;不明
2.臨床症状
歩行障害で発症し、構語障害を伴う口下顎ジストニア、対麻痺、四肢のジストニア、遠位での深部腱反射消失、軸索性ニューロパチー、精神発達遅滞などを認める。緩徐進行性。
3.特記すべき検査所見
(1)脳MRI像:T2画像で淡蒼球の淡明化がみられる。CT所見では淡蒼球の石灰化も指摘されている。
(2)神経病理学所見:鉄沈着は淡蒼球と黒質にみられる。
4.遺伝子診断
COASY遺伝子の病的変異を認める
5.鑑別疾患
ウィルソン病、メンケス病、αfucosidosis、Glutaric aciduria I、リー脳症、神経セロイドリポフスチン症(Neuronal ceroid lipofuscinosis)、ハンチントン病、神経有棘赤血球症、β-hexosaminidase A欠損症、GM1-galactosidase欠損症、ニーマン・ピック病、小児期発症の遺伝性小脳性運動失調、歯状核赤核・淡蒼球ルイ体萎縮症(DRPLA)、家族性痙性対麻痺30型、遺伝性感覚性ニューロパチーⅡC型(HSNⅡC)、遺伝性パーキンソニズム、遺伝性ジストニアに属する疾患群、脳血管障害、抗精神病薬などによる遅発性ジストニア、レット症候群
<無セルロプラスミン血症Aceruloplaminemia/遺伝性セルロプラスミン欠損症(Hereditary ceruloplasmin deficiency)>
1.病因遺伝子と概要
無セルロプラスミン血症(Aceruloplasminemia)は鉄が脳と内臓に蓄積する疾患である。糖尿病(神経所見に10年以上先行することがある)、網膜色素変性症、神経症状を3主徴とする。日本人に多い。
(1)遺伝様式:常染色体潜性遺伝(劣性遺伝)(遺伝子座3q23-q24、原因遺伝子CP、ホモ接合体とヘテロ接合体があり、ほとんどがホモ接合体、遺伝子産物 セルロプラスミン(ceruloplasmin)、MIM ID #604290)
(2)発症年齢:成人期発症(平均51歳)、60~70歳代に死亡する症例が多い。
(3)頻度:5000人、1/2,000,000人(日本)
2.臨床症状
(1)糖尿病
(2)中枢神経症状:神経症状としては認知障害、頭部顔面ジスキネジア、顔面頸部のジストニア、小脳失調が多くの症例で見られる。舞踏運動、パーキンソニズムも頻度が高い。神経症状は30~50歳代に発現することが多い。精神症状としてはうつ、認知障害があり、50歳以上で見られる。
神経所見は様々で小脳失調、姿勢時振戦、舞踏運動-アテトーシスの報告がある。
(3)網膜色素変性症: 93%の症例で見られるが、視力は保たれることが多い。
3.特記すべき検査所見
(1)一般検査所見:糖尿病で本症が発症することが多い(20~40歳代)。コントロール不良例が多い。小球性低色素性貧血を認める。貧血は見かけ上は鉄欠乏貧血であるが、鉄不応性である。糖尿病発症前に貧血が先行することもある。ホモ接合体では血清セルロプラスミンはなく、フェリチン濃度が上昇(正常の12倍程度)をみる。血清セルロプラスミン、フェロキシダーゼ活性は無く、血清銅(10µg/dL以下)、鉄濃度(45µg/dL以下)は低下する。ヘテロ接合体では血清セルロプラスミンレベルは正常の約1/2である。肝臓の鉄濃度は増大する。
(2)脳MRI画像 :T2強調画像で大脳および小脳、淡蒼球、尾状核、被殻、視床、赤核、黒質で低輝度を認め、FDG-PETで発症早期には尾状核の低代謝、進行期には基底核、大脳皮質にまで低代謝領域が広がる。ヘテロ接合体のMRIでは小脳萎縮のみが報告されている。
(3)内臓MRI画像:鉄濃度の増大は内臓でも見られ、肝臓で著しい。
4.遺伝子診断
CP遺伝子の病的変異を認める。
5.鑑別疾患
HFA-associated hereditary hemochromatosis、ウィルソン病、メンケス病、αfucosidosis、Glutaric aciduria I、リー脳症、神経セロイドリポフスチン症(Neuronal ceroid lipofuscinosis)、ハンチントンHuntington病、神経有棘赤血球症、β-hexosaminidase A欠損症、GM1-galactosidase欠損症、ニーマン・ピック病、小児期発症の遺伝性小脳性運動失調、歯状核赤核・淡蒼球ルイ体萎縮症(DRPLA)、家族性痙性対麻痺30型、遺伝性感覚性ニューロパチーⅡC型(HSNⅡC)、遺伝性パーキンソニズム、遺伝性ジストニアに属する疾患群、脳血管障害、抗精神病薬などによる遅発性ジストニア、レット症候群。
<脂肪酸水酸化酵素変異に伴う神経変性症Fatty Acid Hydroxylase-associated neurodegeneration(FAHN):dysmyelinating leukodystrophy and spastic paraparesis with or without dystonia、 spastic paraplegia 35>
1.病因遺伝子と概要
脂質代謝関連酵素由来の脳内鉄沈着神経変性症で、小児期に発症する錐体路障害、小脳性失調/ジストニア、眼症状を示す。進行性に知的機能障害、てんかん発作を生じる。
(1)遺伝様式 常染色体性潜性遺伝(劣性遺伝)(遺伝子座16q21-q23.1、原因遺伝子 FA2H、遺伝子産物 FAHN、MIM ID #612319)
(2)発症年齢:小児期
(3)頻度 1/1,000,000以下
2.臨床症状
古典型と、非典型とがある。3~11歳で発症する錐体路障害(痙性対(四肢)麻痺)、失調/ジストニア、眼症状(視力低下、視野狭窄、色覚障害)を早期に呈する疾患で、進行性に知的機能障害、てんかん発作を生じる。眼球運動系では斜視、側方視眼振、核上性眼球運動障害を認める。7家系の報告があり、対麻痺から痙性四肢麻痺を呈する。構音障害、嚥下障害も示す。
HSP35はFAHNの病型の一つである。
3.特記すべき検査所見
(1)一般検査所見:骨髄でgranular histiocyteを認める。
(2)脳MRI画像所見:T2強調画像で淡蒼球の淡明化を認める。黒質に病巣が波及することは少ない。白質変性と脳梁の非薄化を示す。進行性に小脳半球、虫部、橋、延髄、脊髄の萎縮を認める。
4.遺伝子診断
FA2H遺伝子の病的変異を認める。
5.鑑別疾患
ウィルソン病、メンケス病、αfucosidosis、Glutaric aciduria I、リー脳症、神経セロイドリポフスチン症(Neuronal ceroid lipofuscinosis)、ハンチントン病、神経有棘赤血球症、β-hexosaminidase A欠損症、GM1-galactosidase欠損症、ニーマン・ピック病、小児期発症の遺伝性小脳性運動失調、歯状核赤核・淡蒼球ルイ体萎縮症(DRPLA)、家族性痙性対麻痺30型、遺伝性感覚性ニューロパチーⅡC型(HSNⅡC)、遺伝性パーキンソニズム、遺伝性ジストニアに属する疾患群、脳血管障害、抗精神病薬などによる遅発性ジストニア、レット症候群。
<Kufor-Rakeb症候群>
1.病因遺伝子と概要
脂質代謝関連脳内鉄沈着神経変性症の一つである。Kufor-Rakeb症候群はPARK9(若年発症パーキンソニズムと認知症)とも呼ばれる、neuronal ceroid lipofuscinosisの一つでもある。若年発症のパーキンソニズムで垂直注視麻痺、痙性麻痺を特徴とする。
(1) 遺伝様式;常染色体潜性遺伝(劣性遺伝)(遺伝子座;1p36.13、病因遺伝子;ATP13A2、遺伝子産物;ATPase type 13A2、 OMIM:606693)
(2) 発症年齢;思春期
(3) 頻度:不明
2.臨床症状
若年発症のパーキンソニズムで垂直注視麻痺、痙性麻痺を特徴とする。認知症、錐体路障害、核上性眼球運動障害、顔面・口腔・手指のミニミオクローヌス、視性幻覚、oculogyric crisis、嚥下障害、構音障害を認める。SchneiderらによりNBIAに加えられた。パーキンソニズムにL-dopaは有効であるが、MPANやBPANと同様に早期に運動問題症状を示す。
3.特記すべき検査所見
MRI画像で大脳基底核全体の鉄沈着と全般的な脳萎縮を認める。
4.遺伝子診断
ATP13A2遺伝子の病的変異を認める。
5.鑑別疾患
ウィルソン病、メンケス病、αfucosidosis、Glutaric aciduria I、リー脳症、神経セロイドリポフスチン症(Neuronal ceroid lipofuscinosis)、ハンチントン病、神経有棘赤血球症、β-hexosaminidase A欠損症、GM1-galactosidase欠損症、ニーマン・ピック病、小児期発症の遺伝性小脳性運動失調、歯状核赤核・淡蒼球ルイ体萎縮症(DRPLA)、家族性痙性対麻痺30型、遺伝性感覚性ニューロパチーⅡC型(HSNⅡC)、遺伝性パーキンソニズム、遺伝性ジストニアに属する疾患群、脳血管障害、抗精神病薬などによる遅発性ジストニア、レット症候群。
<Woodhouse-Sakati症候群、別名DCAF17(DDB1 and CLUL4 associated factor17)、Hypogonadism, alopecia, diabetes mellitus, intellectual disability, and extrapyramidal syndrome>
1.病因遺伝子と概要
極めて稀な疾患である。我が国でも症例が確認されている。青年期発症で、多系統性に障害が生じ、多彩な内分泌異常と錐体外路症状、認知機能障害を示す。
(1)遺伝様式;常染色体潜性遺伝(劣性遺伝)(遺伝子座;2q31.1.遺伝子変異;DCAF17(C2orf37)、遺伝子産物;DDBI and CLU4-associated factor 17:DCAF17、OMIM #241080)
(2)発症年齢;青年期
(3)頻度:不明
2.臨床症状
(1)内分泌症状:性腺機能低下、糖尿病、甲状腺機能低下症
(2)神経症状:重度聴覚障害、進行性の錐体外路症状(全身性ジストニアで、顔面ジストニア、舌ジストニアも来す)と精神発達遅滞、認知機能低下を示す。
(3)全身所見:禿頭
3.特記すべき検査所見
(1)一般検査所見:糖尿病、性腺機能低下、甲状腺機能低下、心電図異常
(2)脳MRI所見:淡蒼球と黒質に鉄沈着を認め、白質病巣を伴う。
4.遺伝子診断
DCAF17(C2orf37)遺伝子の病的変異を認める。
5.鑑別疾患
ウィルソン病、メンケス病、αfucosidosis、Glutaric aciduria I、リー脳症、神経セロイドリポフスチン症(Neuronal ceroid lipofuscinosis)、ハンチントン病、神経有棘赤血球症、β-hexosaminidase A欠損症、GM1-galactosidase欠損症、ニーマン・ピック病、小児期発症の遺伝性小脳性運動失調、歯状核赤核・淡蒼球ルイ体萎縮症(DRPLA)、家族性痙性対麻痺30型、遺伝性感覚性ニューロパチーⅡC型(HSNⅡC)、遺伝性パーキンソニズム、遺伝性ジストニアに属する疾患群、脳血管障害、抗精神病薬などによる遅発性ジストニア、レット症候群。
<重症度分類>
Barthel index 85点以下を対象とする。
|
|
質問内容 |
点数 |
|
|
1 |
食事 |
自立、自助具などの装着可、標準的時間内に食べ終える |
10 |
|
部分介助(たとえば、おかずを切って細かくしてもらう) |
5 |
||
|
全介助 |
0 |
||
|
2 |
車椅子からベッドへの移動 |
自立、ブレーキ、フットレストの操作も含む(歩行自立も含む) |
15 |
|
軽度の部分介助又は監視を要する |
10 |
||
|
座ることは可能であるがほぼ全介助 |
5 |
||
|
全介助又は不可能 |
0 |
||
|
3 |
整容 |
自立(洗面、整髪、歯磨き、ひげ剃り) |
5 |
|
部分介助又は不可能 |
0 |
||
|
4 |
トイレ動作 |
自立(衣服の操作、後始末を含む。ポータブル便器などを使用している場合はその洗浄も含む) |
10 |
|
部分介助、体を支える、衣服、後始末に介助を要する |
5 |
||
|
全介助又は不可能 |
0 |
||
|
5 |
入浴 |
自立 |
5 |
|
部分介助又は不可能 |
0 |
||
|
6 |
歩行 |
45m以上の歩行、補装具(車椅子、歩行器は除く)の使用の有無は問わず |
15 |
|
45m以上の介助歩行、歩行器の使用を含む。 |
10 |
||
|
歩行不能の場合、車椅子にて45m以上の操作可能 |
5 |
||
|
上記以外 |
0 |
||
|
7 |
階段昇降 |
自立、手すりなどの使用の有無は問わない |
10 |
|
介助又は監視を要する。 |
5 |
||
|
不能 |
0 |
||
|
8 |
着替え |
自立、靴、ファスナー、装具の着脱を含む |
10 |
|
部分介助、標準的な時間内、半分以上は自分で行える |
5 |
||
|
上記以外 |
0 |
||
|
9 |
排便コントロール |
失禁なし、浣腸、坐薬の取扱いも可能 |
10 |
|
ときに失禁あり、浣腸、坐薬の取扱いに介助を要する者も含む |
5 |
||
|
上記以外 |
0 |
||
|
10 |
排尿コントロール |
失禁なし、収尿器の取扱いも可能 |
10 |
|
ときに失禁あり、収尿器の取扱いに介助を要する者も含む |
5 |
||
|
上記以外 |
0 |
||
※診断基準及び重症度分類の適応における留意事項
1.病名診断に用いる臨床症状、検査所見等に関して、診断基準上に特段の規定がない場合には、いずれの時期のものを用いても差し支えない(ただし、当該疾病の経過を示す臨床症状等であって、確認可能なものに限る。)。
2.治療開始後における重症度分類については、適切な医学的管理の下で治療が行われている状態であって、直近6か月間で最も悪い状態を医師が判断することとする。
3.なお、症状の程度が上記の重症度分類等で一定以上に該当しない者であるが、高額な医療を継続す
ることが必要なものについては、医療費助成の対象とする。
- The NBIA Disorders Association
https://www.nbiadisorders.org/ (英語) - GeneReviews® [Internet]
https://www.ncbi.nlm.nih.gov/books/NBK1141/ (英語) - ジストニア診療ガイドライン2018(日本神経学会監修)
https://www.neurology-jp.org/guidelinem/dystonia_2018.html
治験を実施している指定難病を調べることができます。キーワード検索欄に指定難病名を入力し、検索してください。
治験・臨床研究情報検索サイト一覧